Review of Central Concepts
Before we proceed, it may be beneficial to review some central concepts and themes in the realm of genomics and computational biology. Here we provide a very brief overview of core tenets, and common “gotchas” for these disciplines, as they pertain to this course. We will also introduce the demonstration datasets used throughout the subsequent modules.
Central Dogma
Lets start with the core tenet of genomics, the central dogma:
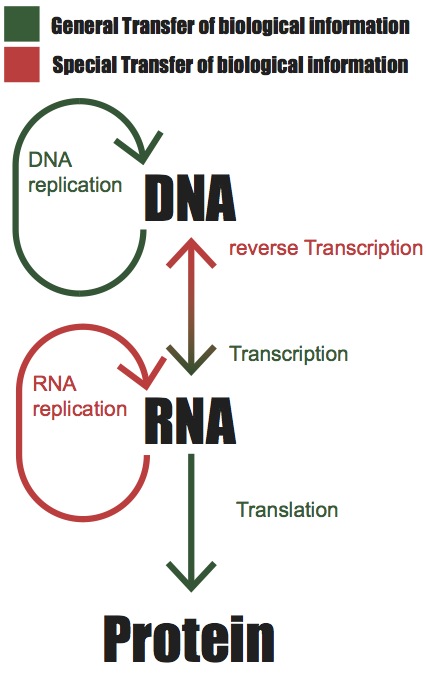
“The Central Dogma. This states that once ‘information’ has passed into protein it cannot get out again. In more detail, the transfer of information from nucleic acid to nucleic acid, or from nucleic acid to protein may be possible, but transfer from protein to protein, or from protein to nucleic acid is impossible. Information means here the precise determination of sequence, either of bases in the nucleic acid or of amino acid residues in the protein.” –Francis Crick 1956
As quoted above, the central dogma describes the flow of biological information, encoded in deoxyribonucleic acid (DNA), ribonucleic acid (RNA) and protein. In essence it states that information cannot flow backward from a protein, that is to say transfer of information from a protein to RNA, DNA, or replicated by another protein is impossible. The general flow of information is through the copying of DNA by DNA replication, the creation of RNA from transcription of DNA, and the creation of proteins via translation of RNA. These processes occur in most cells and are considered mechanisms of general transfer. Special exceptions do exist in the form of RNA replication and reverse transcription however these processes are mostly limited to viruses.
Delving in a bit deeper, DNA takes the form of a double stranded helix comprised of pairs of complementary bases. Adenine (A) and Guanine (G) are classified as purines and complement the pyrimidines Thymine (T) and Cytosine (C) respectively. During transcription DNA is transcribed into a single stranded mRNA molecule in which T bases are converted to uracil (U) and RNA processing such as the removal of introns is performed. This results in a mature mRNA structure composed (for a protein-coding gene) of a 5’ UTR, Coding sequence (CDS), 3’ UTR, and a polyadenylated tail. The mature mRNA is then translated into a peptide sequence beginning at the AUG start codon within the CDS, continuing for each nucleotide triplet codons, until a stop codon occurs. The resulting peptide is often subsequently modified (e.g., phosphorylation of specific residues) and folded into a functional protein. Essentially all “omic” technologies and assays leverage these naturally occuring properties (e.g., DNA/RNA complementarity) and processes (e.g., DNA replication via polymerase). Interpretation of omic data would not be possible without the decades of work to characterize these products, sequence reference genomes (e.g., The Human Genome Project), establish databases of gene annotations, and so on.
What is the reverse complement of: TCGCATTCAGCGGCCATCAGCAGTAAACATCTTAAC?
GTT AAG ATG TTT ACT GCT GAT GGC CGC TGA ATG CGA
Does the resulting sequence contain an open reading frame (ORF)?
Yes. Look for an ATG in frame with a TGA, TAA or TAG, or see the next question for the ORF
What is the mRNA sequence of: ATG TTT ACT GCT GAT GGC CGC TGA?
AUG UUU ACU GCU GAU GGC CGC UGA
What is the translated peptide for the sequence in the previous question?
Met-Phe-Thr-Ala-Asp-Gly-Arg-Stop (Methionine-Phenylalanine-Threonine-Alanine-Aspartic acid-Glycine-Arginine-Stop)
Omic technologies and Data
The advent of rapid and cheap massively parallel sequencing has dramatically increased the availability of genome, transcriptome, and epigenome data. This revolution has given rise to a plethora of standardized (and non-standard) omic analysis workflows, file formats, and a deluge of sequence data. Extracting biologically meaningful conclusions from this data remains a principal challenge. In this course we will learn to visualize several types of omic data in a meaningful way.
Reference Files
Over this course we will be working with a number of reference/data files to aid in analysis and interpretation of our data. All of these files are in standardized formats and are freely available. A brief description of each reference file is given below.
- FASTA: Text files which contain nucleotide or peptide sequences.
- GTF: (Gene Transfer Format) Tab delimited files which hold information regarding gene structure.
- BED: (Browser Extensible Data) Tab delimited files that hold sequence feature information commonly used to diplay data on an annotation track. Coordinates within these files are 0-based.
- VCF (Variant Call Format) Text file used to store observed sequence variations.
- VEP (Variant Effect Predictor) Annotation file used to provide additional information for sequence variations. Originates from the ensembl VEP algortihm.
- MAF (Mutation Annotation Format) Annotation file used to provide additional information for sequence variations. Widely used in The Cancer Genome Atlas project.
- FASTQ Raw sequence file that evolved from FASTA file but that also includes base quality information in a 4 line format.
- SAM/BAM/CRAM Sequence alignment map (SAM) format and it’s compressed equivalents binary (BAM) and compressed reference-based alignment map (CRAM) are files for storing aligned sequencing data. BAM files are the simple binary/compressed equivalent of the SAM file. CRAM uses the reference sequence to more effeciently compress the information in a SAM file. These file types are commonly viewed and manipulated with tools such as samtools, picard, and sambamba to name a few.
What character designates a header in a fasta file?
">"
What character designates a Phred Q value of 30 (Sanger format)?
"?"
For a BAM alignment record, what does a CIGAR string of 60M1D40M signify?
This alignment has 60bp of matching sequence, a one bp deletion, and then an additional 40bp of matching sequence
Genomic annotation resources, browsers, etc.
A common task in genomic analysis and interpretation is collating sequence features or other information to visualize. For example you may have a genomic position and want to know if it is on a gene, the conservation at that position, etc. There are quite a few annotation resources available for this task. In this course we will focus on the ensembl variant effect predictor however a few other resources are listed below.
In the same vein, it is often beneficial to view your reference genome or actual sequence data, again there are many resources available for this, we’ll go over these within the course.
Common problems
Within computational biology there are a number of pitfalls that beginners can fall into. Here we review a few of the most common, to make life easier down the road.
Genomic coordinate systems
Within computational biology there are two competing coordinate systems for specifying regions within the genome. These two systems number either the nucleotides in the genome directly (1-based), or the gaps between nucleotides (0-based). Let’s look at the diagram below displaying an imaginary sequence on chromosome 1 to help illustrate this concept.
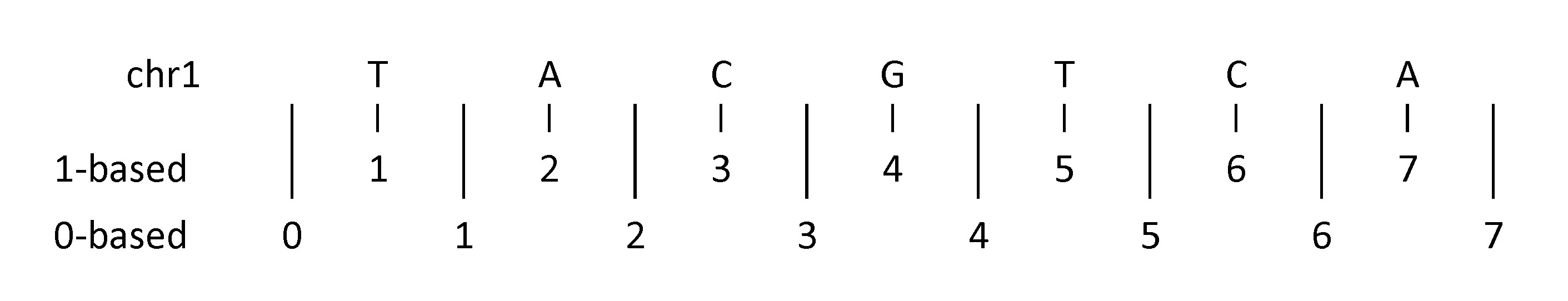
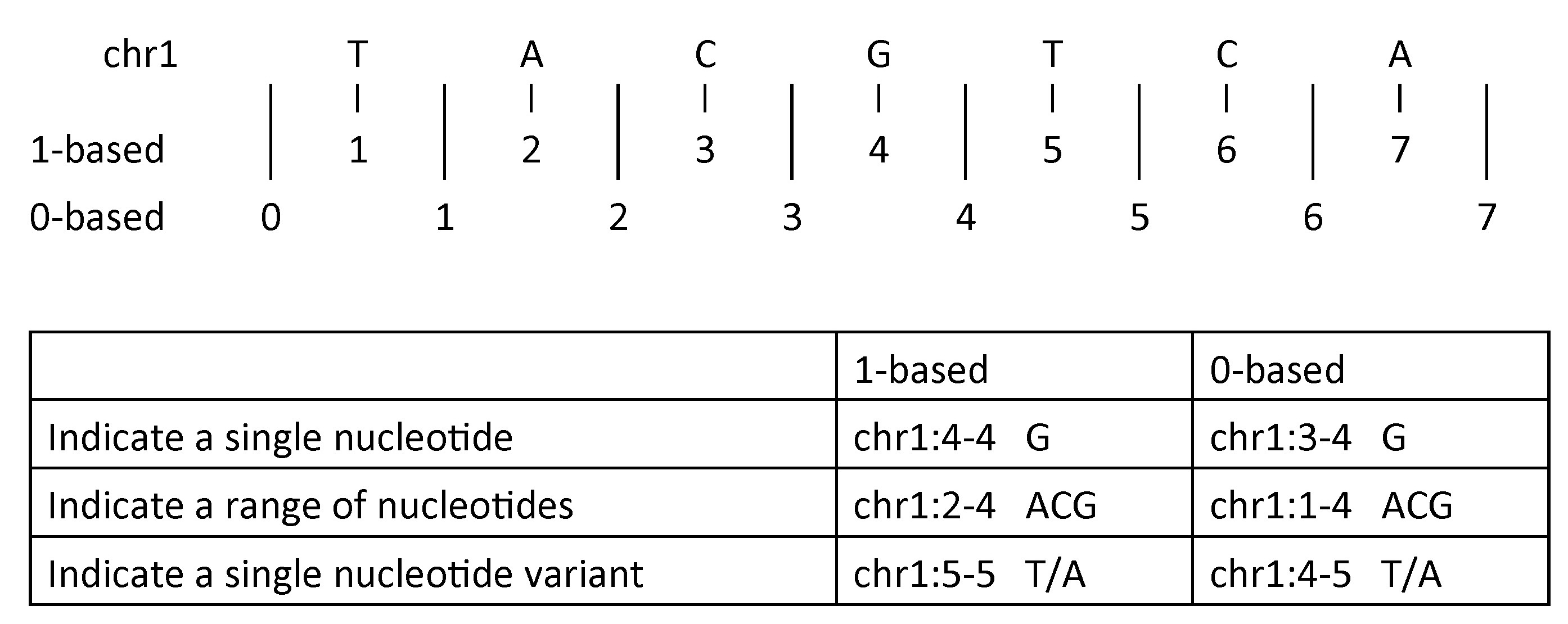
To indicate a single nucleotide variant:
- 1-based coordinate system
- Single nucleotides, variant positions, or ranges are specified directly by their corresponding nucleotide numbers
- 0-based coordinate system
- Single nucleotides, variant positions, or ranges are specified by the coordinates that flank them
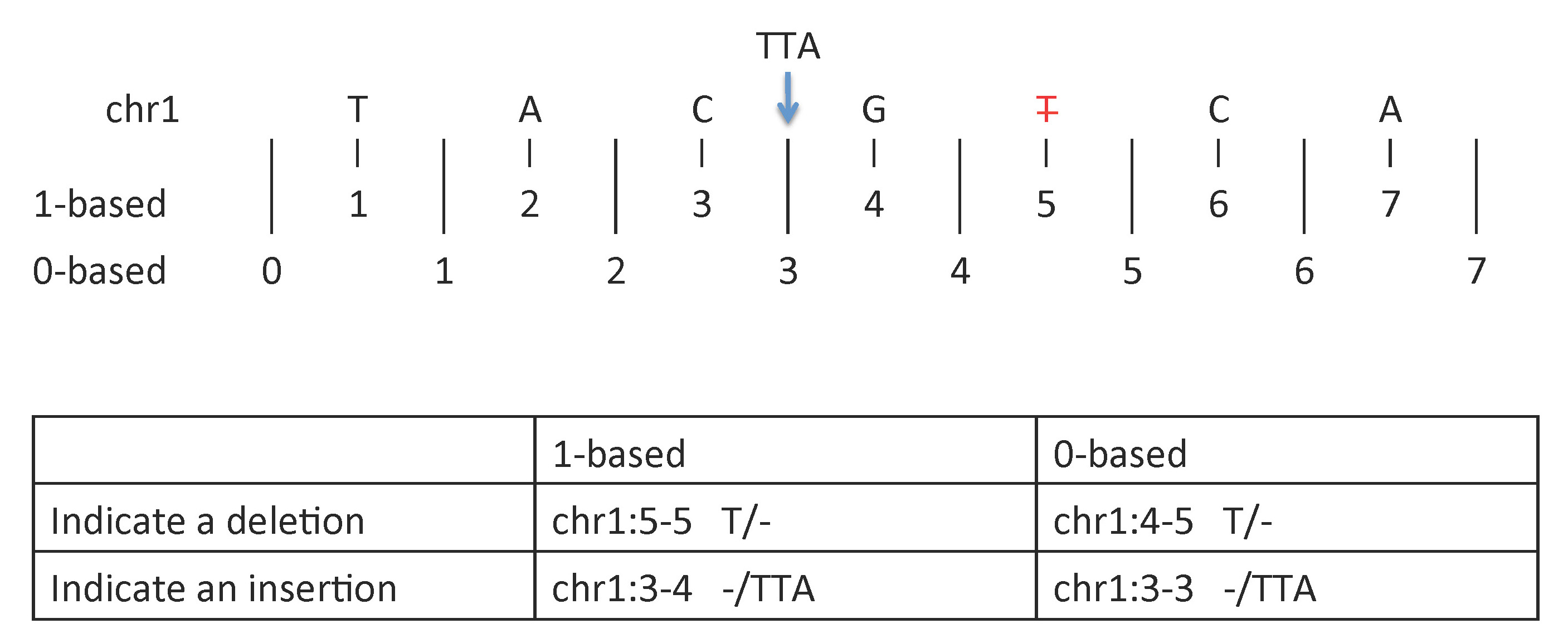
To indicate insertions or deletions:
- 1-based coordinate system
- Deletions are specified directly by the positions of the deleted bases
- Insertions are indicated by the coordinates of the bases that flank the insertion
- 0-based coordinate system
- Deletions are specified by the coordinates that flank the deleted bases
- Insertions are indicated directly by the coordinate position where the insertion occurs
What is the difference between 0-based and 1-based coordinates?
0-based coordiantes number between nucleotides, 1-based coordinates number nucleotides directly.
Differences in carriage returns
In all unix based systems (OSX included) new line characters, commonly referred to as carriage returns are designated by the character “\n”. However this is not a universal standard, windows programs such as Excel designate a carriage return as “\r\n” mostly to maintain compatability with MS-DOS. This can create problems when attempting to use a file made via a windows application on a unix system. Specifically, attempting to view one of these types of files on a unix system will not interpret “\r\n” as a new line but rather as “^M”. This is something to be aware of but is fortunately easily remedied through any text processing language. Below you will find an example for fixing this file through perl via a command line.
perl -pi -e 's/\r\n|\n|\r/\n/g' FileToChange.txt
In essence the command above calls the perl regular expression engine and substitutes “\r\n” or “\n” or “\r” with “\n”, editing the file in place.
Genome builds
When doing any sort of bioinformatic analysis it is good to be aware of the reference assembly upon which your data is based. Each species has it’s own reference assembly representing the genome of that species, and each species specific assembly can have multiple versions as our understanding of the genome for each species improves. When comparing across data sets, especially in terms of genomic coordinates, reference assemblies should always match. We will cover all of this in more detail in the liftOver section.
A detailed discussion of some commonly used human reference genome builds can be found here.
Introduction to demonstration data sets
Throughout this course we will be working with and visualizing many different datasets. Below we provide a brief overview of each core data set and what type of visualizations we will create with them.
- Somatic variant calls from the manuscript “Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma”
- General ggplot2 graphs
- Visualizing transitions and transversions
- Interactive visualizations with shiny
- Visualization of sequencing coverage
- Somatic variant calls from the manusctipt “A Phase I Trial of BKM120 (Buparlisib) in Combination with Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer.”
- Waterfall plots
- Copy Number data from Copycat2 related to the manuscript “Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial.”
- Various copy number visualizations
- Aligned bam files and varscan output files for the HCC1395 breast cancer cell line
- Visualizing data with IGV
- Loss of Heterozygosity graphics
- Expression data from the manuscript “A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients.”
- Differential expression
- Pathway analysis
- Pathway Visualization
- Coverage data from bedtools related to the manuscript “Truncating Prolactin Receptor Mutations Promote Tumor Growth in Murine Estrogen Receptor-Alpha Mammary Carcinomas”
- Displaying gene coverage