ClinVar
ClinVar aggregates information about genomic variation and its relationship to human health. To date, thousands of variants and associated phenotypes have been deposited in ClinVar. The main use case for ClinVar is when you have one or more variants observed in a human sample and you would like to know if these variants have established clinical relevance. The resource is fairly heavily biased towards germline inherited variants that predispose an individual for disease. Many diseases and phenotypes have been reported. Some of the variant observations in ClinVar come from sources that mine published literature (e.g. DoCM, OMiM, etc.). However, the majority of submissions to ClinVar come from clinical sequencing labs that identify variants in patients and report that variant along with the phenotype of the patient(see Data Sources for more details). Several types of variants are supported in ClinVar. Many are single nucleotide variants (SNVs) or small insertions and deletions, but large structural variants are also supported. In general, all variants in ClinVar are designated using the HGVS nomenclature. ClinVar strongly recommends following the ACMG guidelines in deciding on the clinical significance (pathogenicity) of variants. The following tutorial explores these concepts in greater detail.
Basic intro to the ClinVar interface
ClinVar offers a few video tutorials to get you started: Intro to ClinVar and Find All Variants with ClinVar. The ClinVar website is quite simple. The home page offers various links to frequently used sections. The tool bar at the top of each page includes the simple search bar and various menus to navigate to additional information.
- To get a sense of where the data in ClinVar is coming from, visit the complete submitter list (Statistics -> List of Submitters). This list gives you an idea of some of the major players in the genetic diagnostics and variant interpretation.
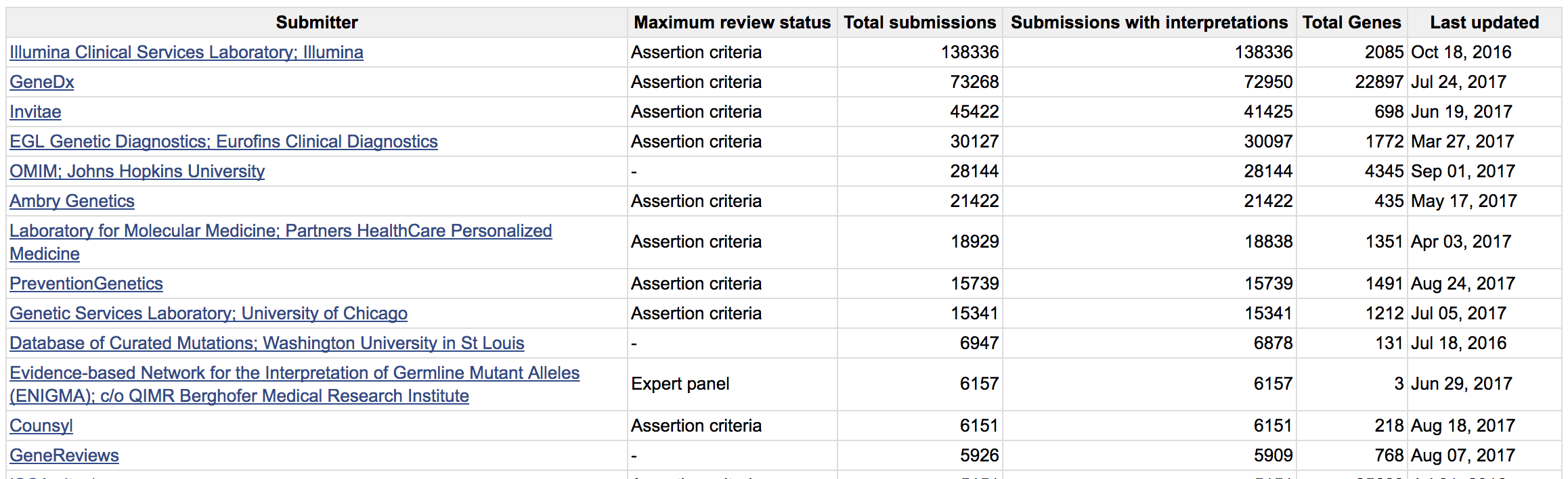
- To get a sense of the current content of ClinVar, visit the Statistics page (Statistics -> Statistics).
- If you would like to download raw ClinVar data, you can do so from their FTP site. For example, you can download the complete set of ClinVar variants in VCF format (human genome build 37 VCF or human genome build 38 VCF).
ClinVar has several key entities used to organize variant data. (1) Each submission of an interpretation for a variant is assigned a submission accession of the format SCV000000000.0. Multiple labs could report independent interpretations for the same variant. Each would be entered as a distinct record. If a lab wishes to update that submission, they can do so, and the version number will be updated to reflect this change. (2) When there are multiple submissions about the same variation/condition relationship, they are aggregated under a reference accession of the format RCV000000000.0. If the same variant is associated with multiple conditions, there will be multiple reference accessions. (3) All information about a single distinct variant are organized under one Variation ID and a corresponding Allele ID. Both are simple integers. Each allele corresponds to a distinct genomic variation that must be described using one or more HGVS expressions.
Consider the following example: NM_007294.3(BRCA1):c.5558dupA (p.Tyr1853Terfs)
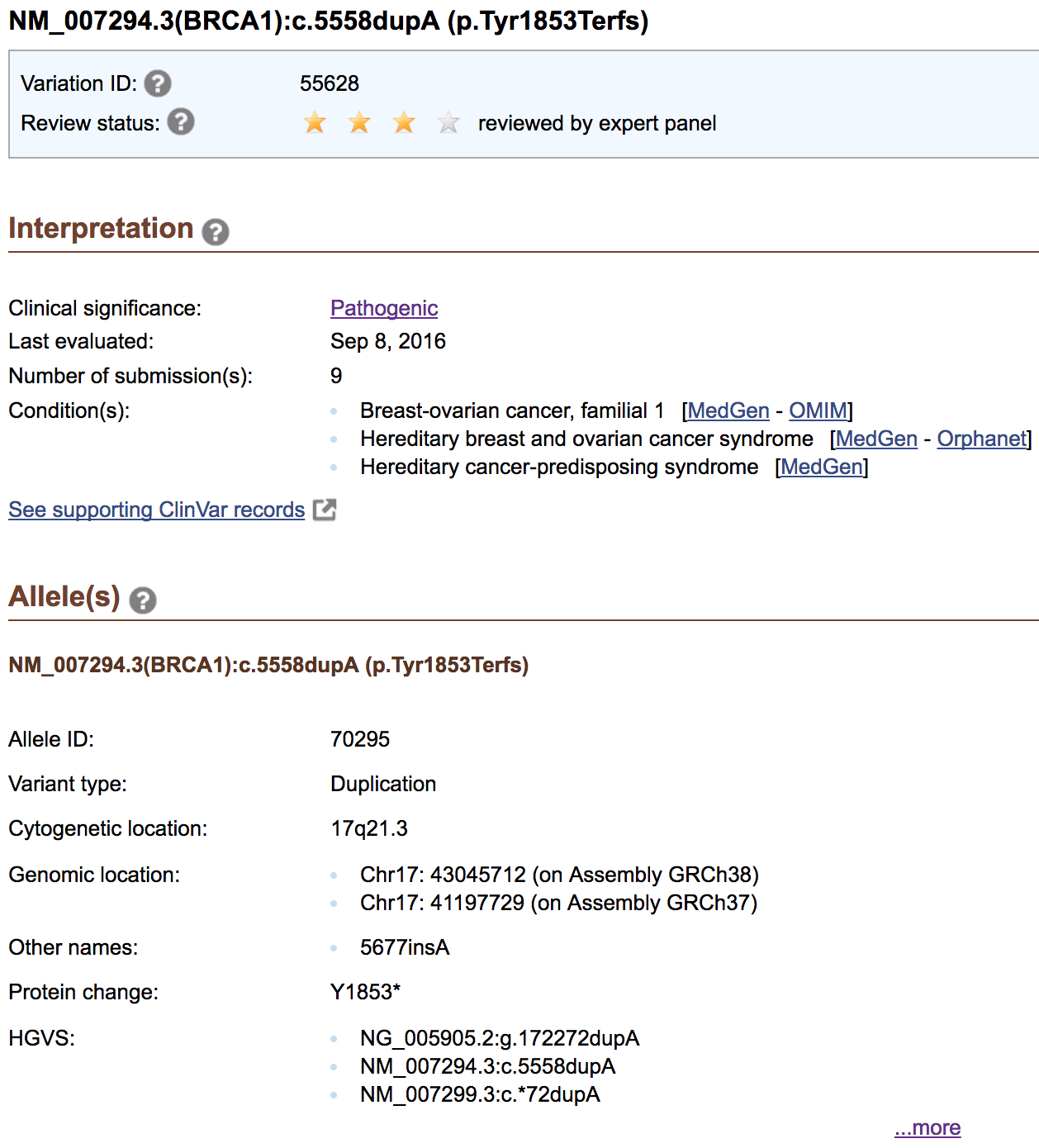
- Variation ID: 55628
- Allele ID: 70295
- Submission accessions: SCV000300281.2, SCV000329130.4, SCV000488476.1, …
- Reference accessions: RCV000074357 (Breast-ovarian cancer, familial 1), …, RCV000163976 (Hereditary cancer-predisposing syndrome)
A more detailed explanation of these identifiers can be found in the Identifiers section of the ClinVar documentation.
Searching ClinVar
ClinVar several search modes. (1) you can simply type free form text in the search box near the top of every page, (2) if you know the neccessary field codes, you can construct complex queries in this same search box, (3) you can use the Advanced Search Builder.
- Use the basic search box to find all variants for the gene AKT1. Note that when ClinVar detects a match to a proper gene name in your search, it assumes you want to search against only the gene name field. It indicates this by adding [gene] to your search text. If you wish to search all fields for this term, you can can chose the option to “Search instead for all ClinVar records that mention AKT1”.
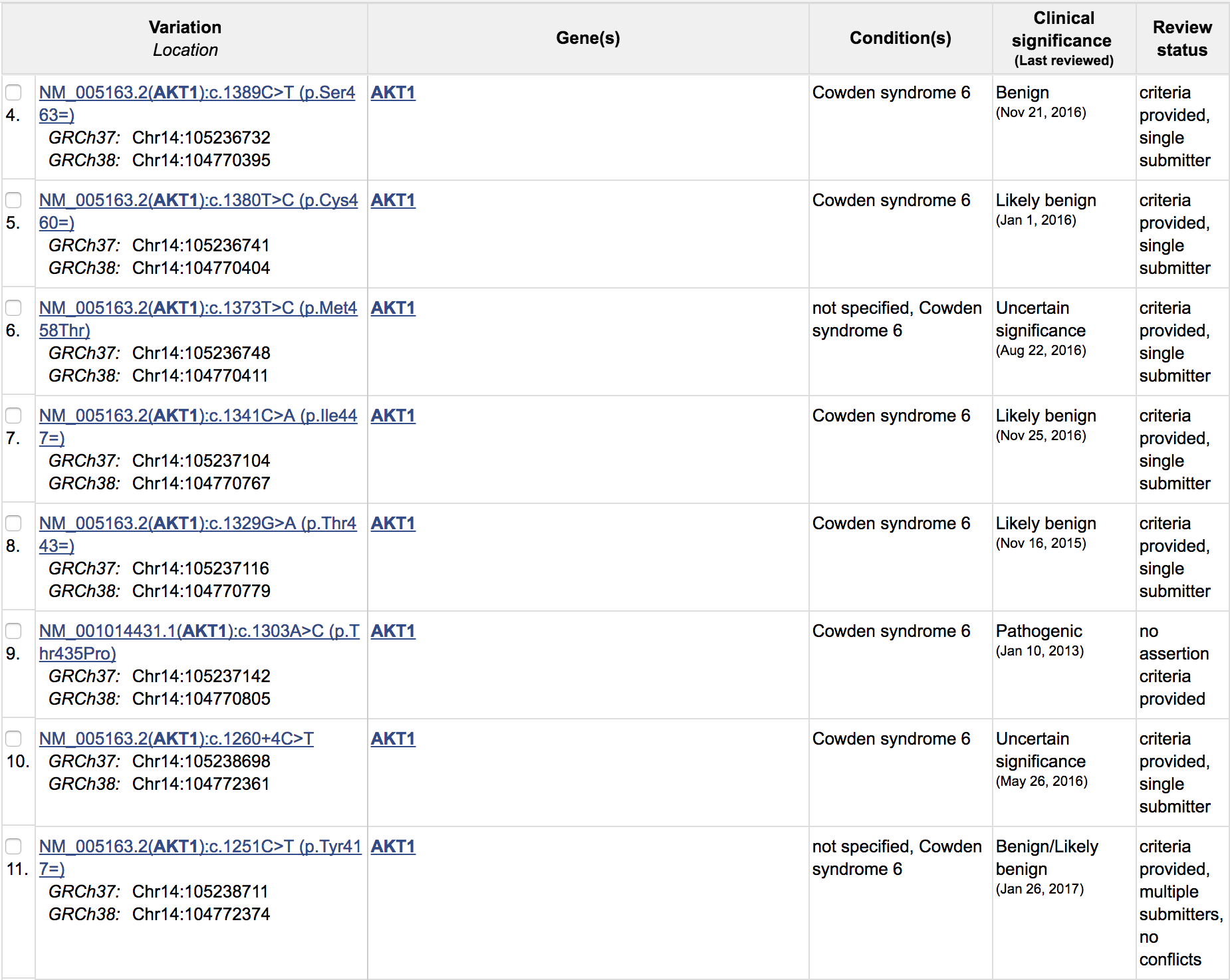
- Now try searching for a specific disease: proteus syndrome. What is the popular significance of this disease? Note that this search appears to be returning partial matches to various conditions. To get a more refined search, trying placing the condition name in quotes and adding the disease tag: “proteus syndrome”[dis]. Remember that we can find possible tags to use here on the help page. Note the variant NM_005163.2(AKT1):c.49G>A (p.Glu17Lys). This variant has significance in both the germline and somatic contexts. In the germline context, it appears pathogenic for Proteus Syndrome (actually in this case, this involves somatic mosaicism).
-
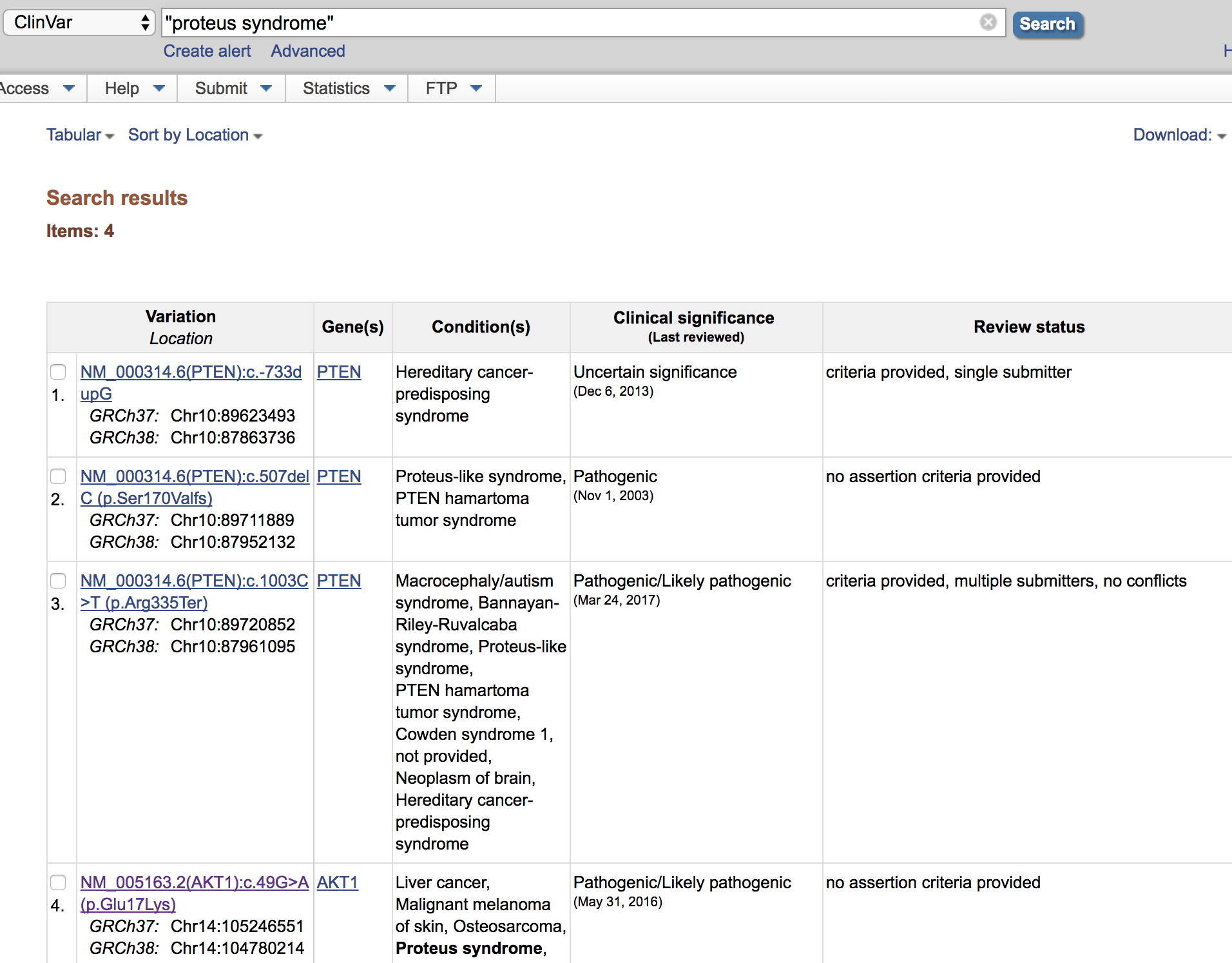
Search for variants within a narrow window on a single chromosome: “5[chr] AND 1264532:1264552[chrpos]”. What gene does this region correspond to? What is the significance of this gene?
-
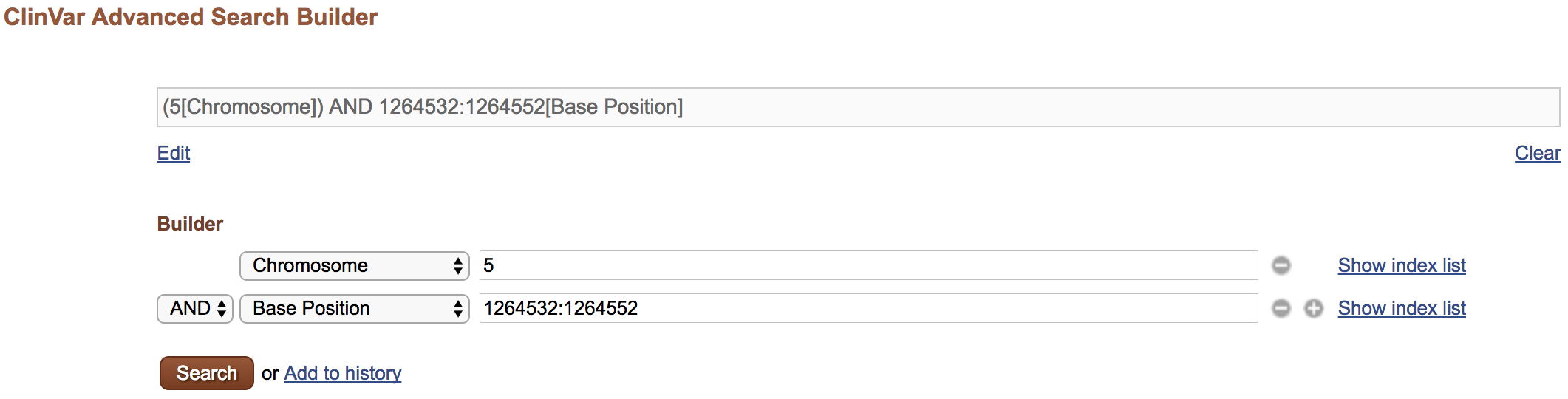
Use the Advanced Search page to construct the same query. Note that you can use the show index list option to see valid search options/examples for each field.
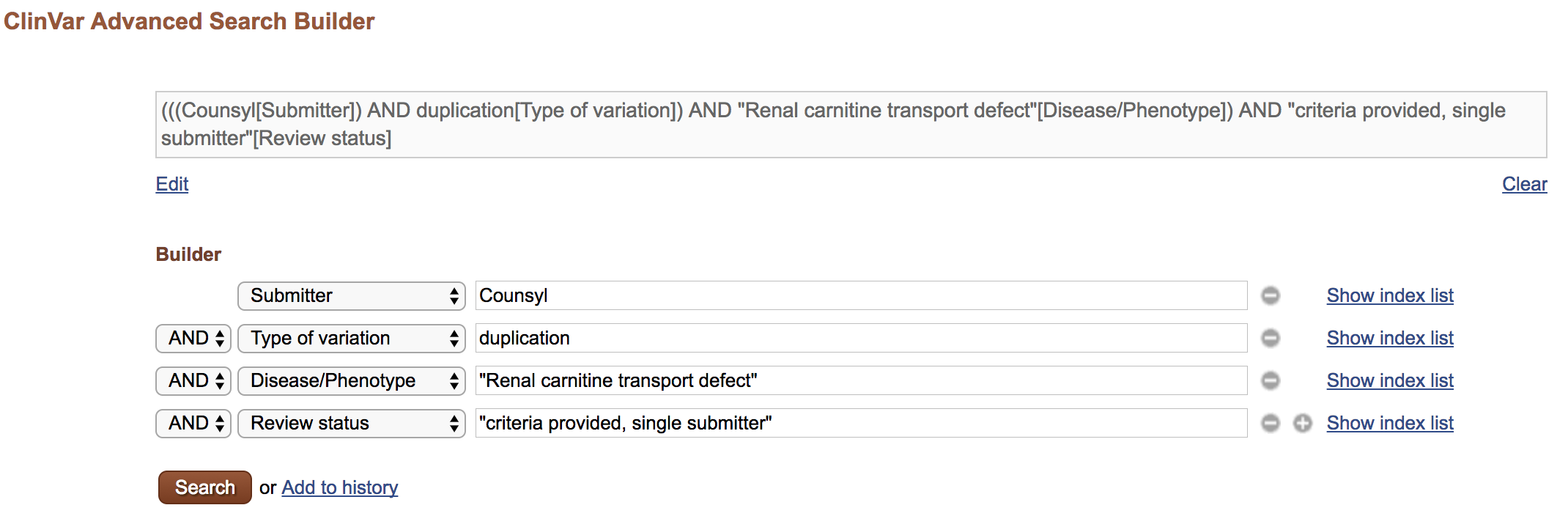
- Use the Advanced Search page to construct a complex query. For example: Variants submitted by “Counsyl, that are “duplication variants”, for the disease “Renal carnitine transport defect”, and with review status “criteria provided, single submitter”.
Obtaining a list of all high quality predisposing variants in ClinVar
If you just want the complete list of high quality variants that you might consider returning results for in a clinical setting. You might decide to peform a query that requires the variant to have been reviewed by an expert panel, belong to a practice guideline, or have been submitted by multiple entities that agree on the interpretation. This list could be further limited to those that are Pathogenic and where the variant origin is stated as “germline”. If you put all of this together the query looks like this:
((("reviewed by expert panel"[Review status]) OR "criteria provided, multiple submitters, no conflicts"[Review status]) OR "practice guideline"[Review status]) AND "clinsig pathogenic"[Properties] AND "germline"[Origin]
- Download this complete data set to a tabular (TSV) file and open it is a spreadsheet editor such as Excel.
A brief primer on HGVS
In completing the exercises above in ClinVar you will have noticed that variants have rather complex looking names. This is a deliberate choice on the part of ClinVar. It is common in the literature and in day to day conversations to refer to variants with shorthand names. For example, someone refers to BRAF V600E you may have a good idea what they are referring to. However, such names can be ambigiguous. There are often multiple correct variations in the genome that could lead to a V600E effect in the protein. Furthermore, if you want to design an assay to detect this variant in RNA, cDNA, or genomic DNA, then V600E is not very useful. For this reason, the community has adopted a set of standards for describing variants in a much more precise way that is unambiguous and leads to less confusion. The Human Genome Variation Society (HGVS) acts as a steward for these standards. They have also released detailed guidelines (the rulebook) on the correct way to create HGVS expressions for almost any variant. These guidelines are maintained, updated, and versioned. They are available at: varnomen.hgvs.org. ClinVar also provides a simple overview of the HGVS types they use. Here is an example of the same variant (AKT G17K) represented as HGVS expressions describing it at the:
ClinVar variant name: NM_005163.2(AKT1):c.49G>A (p.Glu17Lys) HGVS for protein: NP_005154.2:p.Glu17Lys HGVS for cDNA: NM_005163.2:c.49G>A HGVS for genome: NC_000014.9:g.104780214C>T (GRCh38)
Each HGVS has several required elements. First you have a sequence accession with a version number (e.g. NM_005163.2). This tells you unambiguously what the reference sequence is (a published protein, cDNA, or genome sequence). Then you have a delimiter “:”. Next you have a letter that indicates the general type of HGVS expression to follow: “p” for protein, “c” for cDNA, “g” for genomic DNA. Finally, you have the expression that describes the sequence variation itself. The rules for these expressions depend on the type of variation (substitution, duplication, deletion, etc.) and the level of HGVS (protein, cDNA, gDNA, RNA, …).
- As an exercise. Try manually creating valid HGVS expressions at the genome, cDNA, and protein level for: BRAF V600E. What fundamental tools would we need to do this (assuming we can not just look it up in a database)?
Here are some excellent resources for working with HGVS:

- Mutalyzer (tools for validating and converting HGVS expressions)
- TransVar (tool for converting HGVS expressions)
- MyVariantInfo (aggregates variant info including HGVS expressions from many sources)
A brief primer on the ACMG guidelines for germline variant interpretation
You may have noticed in the exercises above various references to the clinical significance or pathogenicity of variants. In some queries of ClinVar we limited variants to only those that are thought to be Pathogenic (i.e. if inherited, the predispose an individual to a particular disease). How does one establish whether a variant is Pathogenic, or Benign? To help establish how this should be done, the ACMG has released detailed Standards and guidelines for the interpretation of sequence variants. For the full details, one should definitely read that paper and refer back to it extensively as you try to interepret specific variants. Briefly, the ACMG established a variant classification approach that is meant to be applicable to variants in all Mendelian genes. They categorize the types of evidence that one should use to support the pathogenicity of a variant. Each type of evidence is indicated by an evidence code. Evidence codes are broken down into two main categories, those that support a pathogenic interpretation, and those that support a benign interpretation. Within these two broad groups, sub-categories are defined for different types of evidence (e.g. functional data, population frequency, inheritance patterns, algorithmic predictions, etc.). The guidelines describe how to document this evidence and how to use that evidence to make a final conclusion for the pathogenicity of each variant for each disease/phenotype. The ACMG defines five tiers for the pathogenicity of a variant: “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign”.
We strongly encourage you to read the ACMG guidelines carefully. The critical summaries are provided in several tables of this paper.

- Evidence and codes for classifying pathogenic variants: Table 3
- Evidence and codes for classifying benign variants: Table 4
- Rules for combining evidence to classify sequence variants: Table 5
Final comment: If used properly, the ACMG guidelines require a lot of evidence to define a variant as pathogenic or even likely pathgenic. Similarly strong evidence is needed to conclude a variant is benign or even likely benign. Many variants are thus initially classified as “uncertain significance”. Note that this does not mean unknown it means uncertain. i.e. lacking certainty. Since variant classifications are meant to be used in a clinical setting the goal is to avoid misinforming patients that may take life changing actions upon hearing these results.
ClinVar practice exercises
How many ClinVar variants are there for BRCA2 that are Germline, Pathogenic and have Expert Panel review status?
Get a hint!
Simply enter BRCA2 in the ClinVar search box, then apply the three filters requested.
Answer
At the time of writing this post, there were 2069 germline, pathogenic, expert panel reviewed BRCA2 variants.
Use ClinVar to find valid HGVS expressions at the genome, cDNA, and protein level for the following variant: PIK3CA H1047R.
Get a hint!
Simply enter PIK3CA H1047R in the ClinVar search box, then review the HGVS section.
Answer
NG_012113.2:g.90775A>G (genome), NM_006218.3:c.3140A>G (cDNA), NP_006209.2:p.His1047Arg (protein).
Find all of the variants in ClinVar that correspond to exon 2 of VHL. How many are there? Export these to a spreadsheet.
Get a hint!
Use a genome browser such as IGV to obtain the build38 coordinates for exon 2 of VHL. Then use the advanced ClinVar search box to search for variants in VHL and within the exon 2 coordinates.
Answer
To get VHL exon 2 variants use this query. (VHL[Gene Name]) AND 10146514:10146636[Base Position]. At the time this question was created there were 517 variants in VHL and 63 of these were within exon 2.