Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
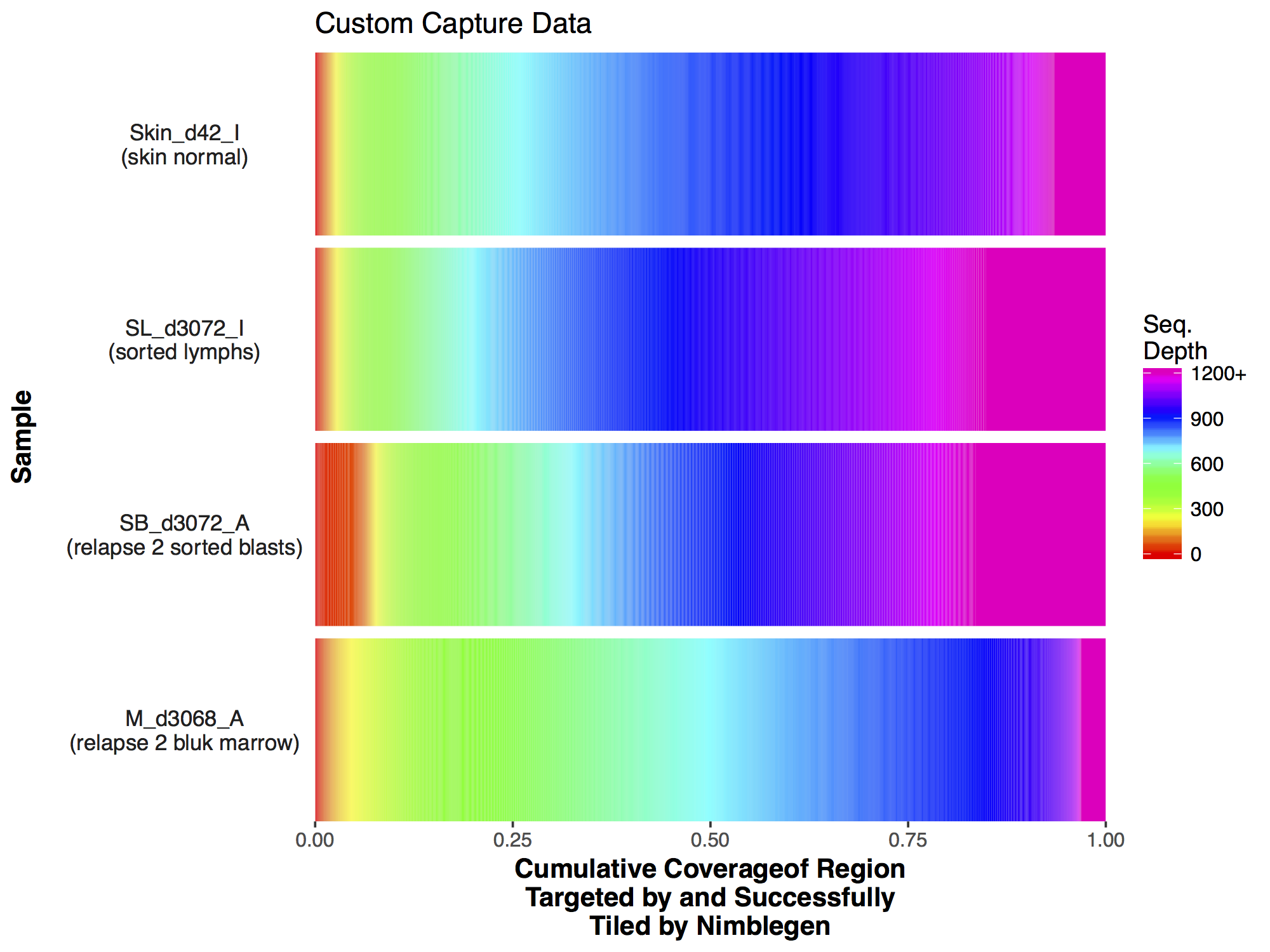
Commonly when a sample has undergone sequencing you will want to know the sequencing depth achieved in order to get an idea of the data quality. The covBars() function from the GenVisR package is designed to help in visualizing this sort of data by constructing a color ramp of cumulative coverage. In this section we will be reconstructing the capture coverage plots for 4 samples from the paper “Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia” shown in Supplemental Figure S3.
The covBars() function takes as input a matrix with rows representing the sequencing depth, columns representing samples, and matrix values representing the number of reads meeting that criteria for a given cell. The best way to begin constructing this data is with the command line tool samtools depth for anything other than whole genome sequencing data in which case picards CollectWgsMetrics program might be a better choice. The output from samtools depth for 4 capture samples from the adult B-lymphoblastic leukemia manuscript linked above is available on genomedata.org. The command used to create this file was samtools depth -q 20 -Q 20 -b roi.bed -d 12000 sample1.bam sample2.bam sample3.bam sample4.bam, let’s briefly go over the parameters used to create this file and load it into R. Within the samtools depth command we used the parameters -q 20 and -Q 20 to set a minimum base and mapping quality respectively. Essentially this means that a read will not be counted if these criteria are not met. We used -b roi.bed to tell samtools depth to only create pileups for those regions encompassed by each line of the bed file. We used -d 12000 to ensure pileups are not stopped until 12,000 reads are reached for that coordinate. Finally we specified the four bam files for which we want to create pileups for, each bam file has been indexed with samtools index.
# load the ouput of samtools depth into R
seqData <- read.delim("http://genomedata.org/gen-viz-workshop/GenVisR/ALL1_CaptureDepth.tsv", header=F)
colnames(seqData) <- c("chr", "position", "samp1", "samp2", "samp3", "samp4")
seqData <- seqData[,c(3:6)]
After reading in the data you’ll notice that the format is not quite what covBars() will accept; Instead of having read pileup counts for each coverage value (1X 2X 3X etc.) we have read pileups for each coordinate position in the bed file. Let’s go step by step how to manipulate the data into the proper format for covBars(). The first thing we need to do loop over all columns of seqData and obtain a tally of read pileups for each coverage value, this can be done with a call to apply() using the plyr function count(). Strictly for readability we then create our own function called renameCol with the purpose of renaming the columns names to a more human readable format and use lapply() to apply the function to each data frame in our list. You might notice that each data frame in seqCovList has a differing number of rows. This has occurred because not every sample will have pileups for the same coverage values, this is especially true when getting into higher coverage depths owing to outliers. We will fix this by creating a framework data frame containing all possible coverage values, from the min to the max observed from each data frame in the list seqCovList. For this we use lapply() to apply max() to each coverage column within the data frames in seqCovList using an anonymous function within the lapply(). This will return a list of the maximum coverage value for each data frame so we use unlist() to coerce the list to a vector and use max() again to find the overall maximum coverage observed. A similar procedure is perormed to obtain the minimum coverage and we create are framework data frame using these maximum and minimum values with data.frame(). Now that we have our framework data frame we can use merge() and lapply() to perform a natural join between the the framework data frame covFramework and each data frame containg the acutal read pileups for each coverage in seqCovList. From there we can use Reduce to recursively apply merge() to our previous list of data frames seqCovList, which effectively merges all data frames in this list. Our data is now in a format covBars() can accept but we have to do some minor cleanup. The merges we performed introduce NA values for those cells which had no coverage pileups, we convert these cell values from NA to 0. We then remove the coverage column from the data frame as that information is defined in the row names, convert the data frame to a matrix, and add in column names.
# Count the occurrences of each coverage value
# install.packages("plyr")
library(plyr)
seqCovList <- apply(seqData, 2, count)
# rename the columns for each dataframe
renameCol <- function(x){
colnames(x) <- c("coverage", "freq")
return(x)
}
seqCovList <- lapply(seqCovList, renameCol)
# create framework data frame with entries for the min to the max coverage
maximum <- max(unlist(lapply(seqCovList, function(x) max(x$coverage))))
minimum <- min(unlist(lapply(seqCovList, function(x) min(x$coverage))))
covFramework <- data.frame("coverage"=minimum:maximum)
# Merge the framework data frame with the coverage
# seqCovList <- lapply(seqCovList, function(x, y) merge(x, y, by="coverage", all=TRUE), covFramework)
seqCovList <- lapply(seqCovList, merge, covFramework, by="coverage", all=TRUE)
# merge all data frames together
seqCovDataframe <- Reduce(function(...) merge(..., by="coverage", all=T), seqCovList)
# set all NA values to 0
seqCovDataframe[is.na(seqCovDataframe)] <- 0
# set the rownames, remove the extra column, and convert to a matrix
rownames(seqCovDataframe) <- seqCovDataframe$coverage
seqCovDataframe$coverage <- NULL
seqCovMatrix <- as.matrix(seqCovDataframe)
# rename columns
colnames(seqCovMatrix) <- c("sample1", "sample2", "sample3", "sample4")
Now that we have our matrix we can simply call covBars() on the resulting object. The output looks significantly different from the manuscript version though so what exactly is going on? The problem is with the coverage outliers in the data, the graph shows that 99% of the genome is covered up to a depth of 2000X however are scale is linear and so the outliers are forcing are scale to cover a range from 0 to 11,802, obviously this is problematic.
# run covBars
covBars(seqCovMatrix)
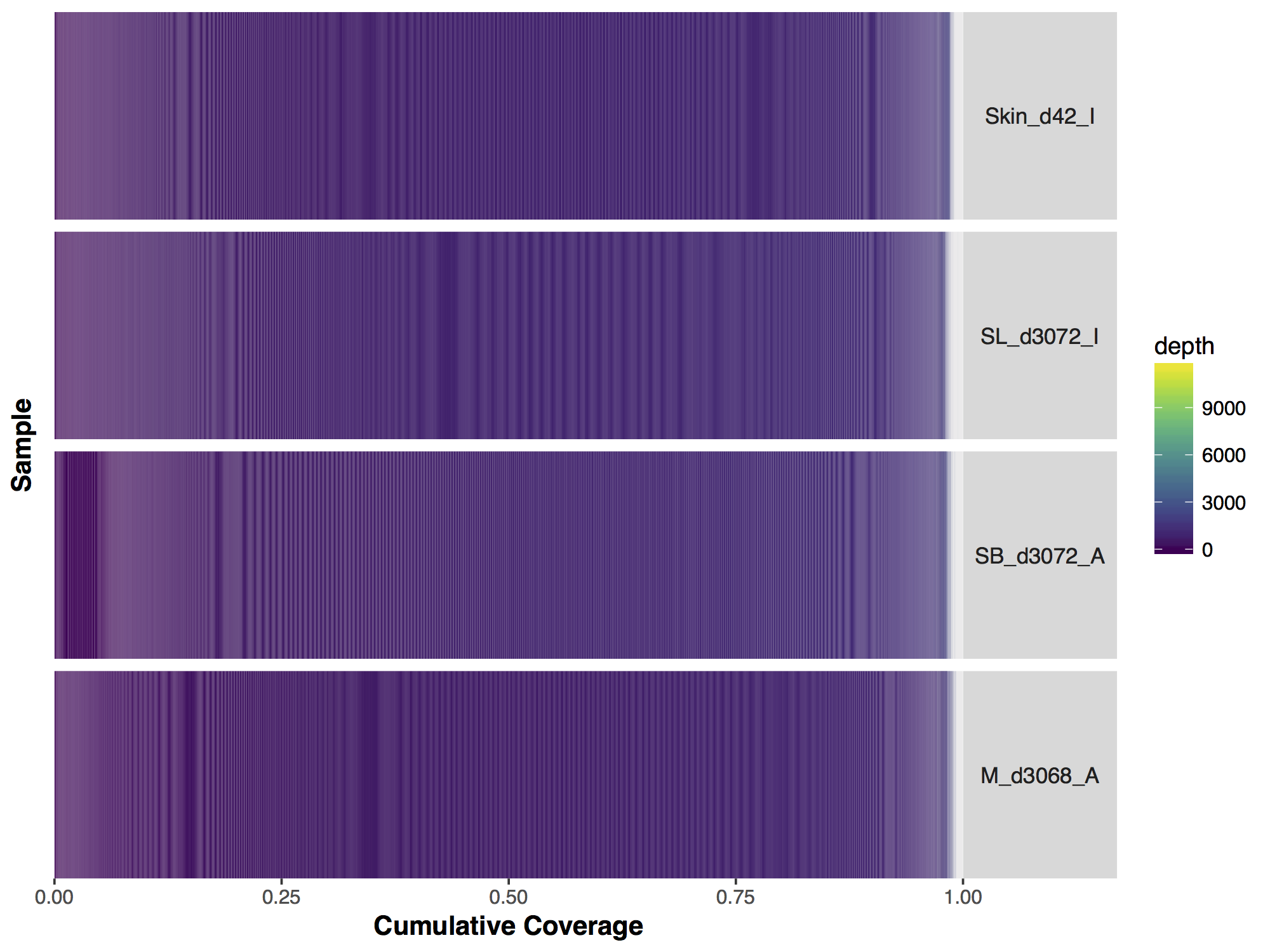
If we look closely at the manuscript figure we can see that the scale is actually limited to a coverage depth of 1,200. This ceilings the outliers in the data and puts everything on an easily interpretable scale. Let’s go ahead and do this to our data; First we need to calculate the column sums for every coverage value (i.e. row in our data) beyond 1,200X, for this we use the function colSums(). The previous function returns a vector, we convert this to a matrix with the appropriate row name 1200. From there we subset our original matrix up to a coverage of 1,199X and use rbind() to add in the final row corresponding to a coverage of 1,200X.
# ceiling pileups to 1200
column_sums <- colSums(seqCovMatrix[1200:nrow(seqCovMatrix),])
column_sums <- t(as.matrix(column_sums))
rownames(column_sums) <- 1200
seqCovMatrix2 <- seqCovMatrix[1:1199,]
seqCovMatrix2 <- rbind(seqCovMatrix2, column_sums)
# run covBars
covBars(seqCovMatrix2)
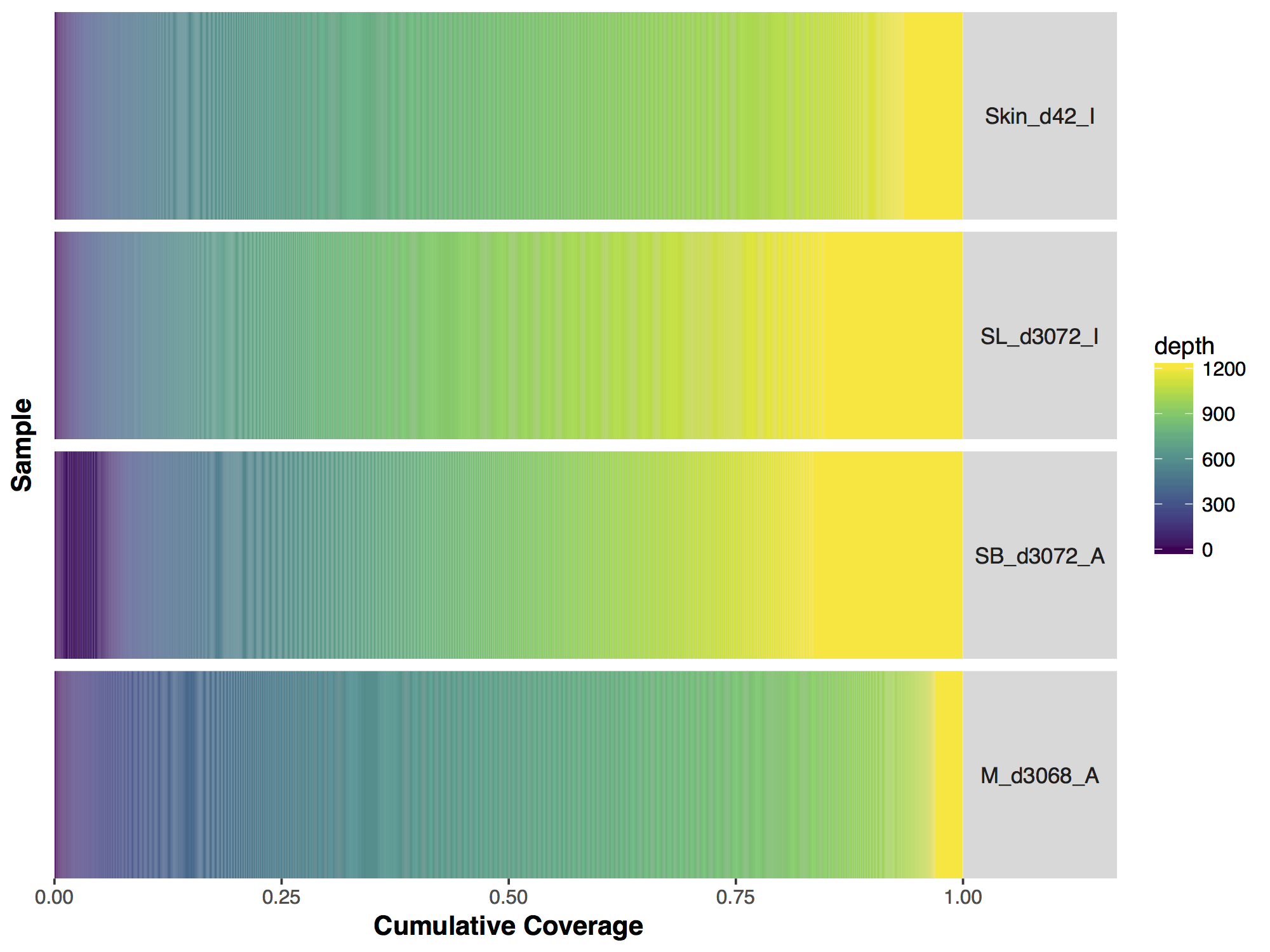
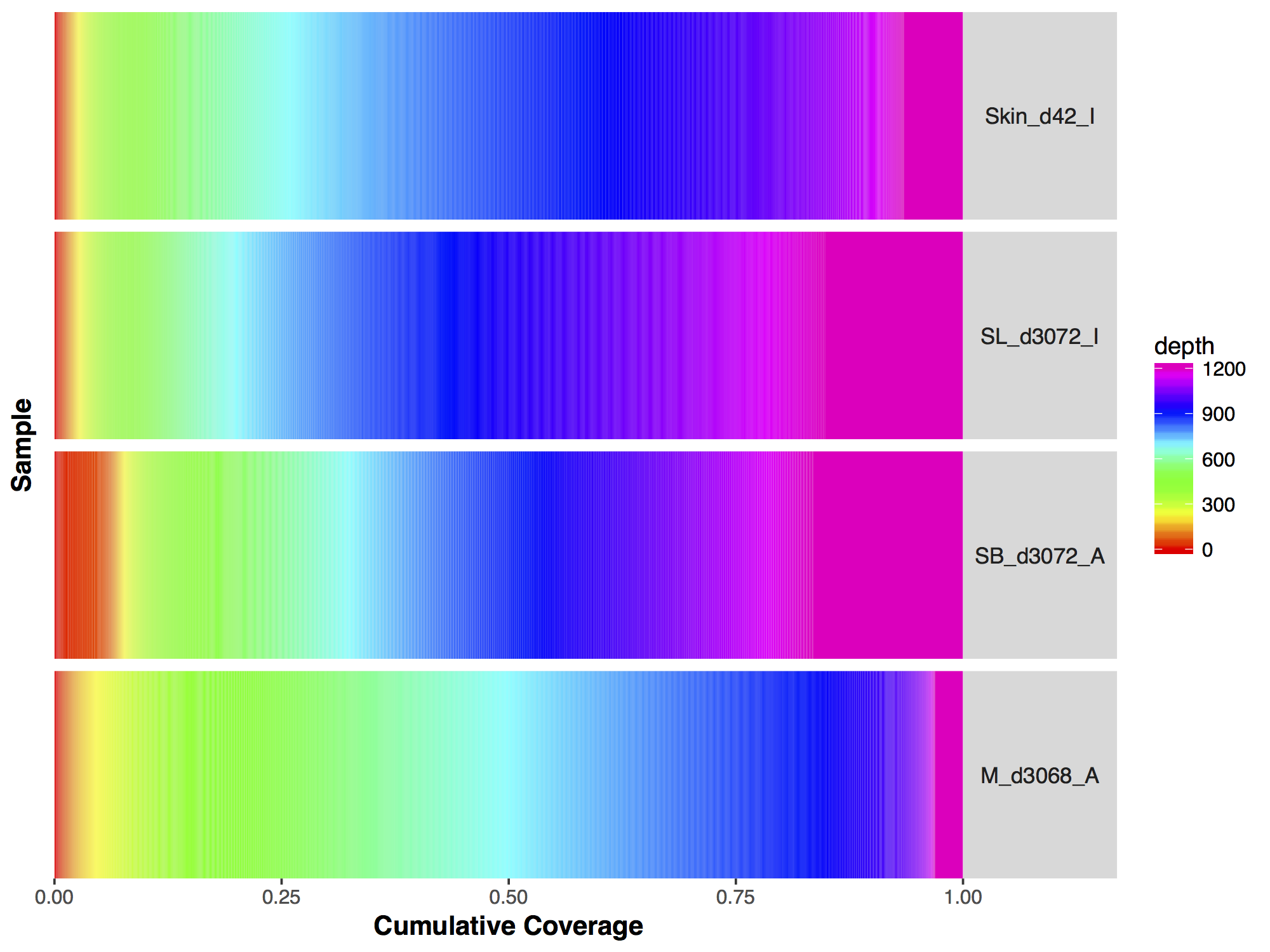
Now that we have a descent scale let’s change the colours within the scale to more closely match that of the figure. Adding more colours in our colour ramp will also provide more resolution for our interpretation of the data. For this we can use the function rainbow() and subset the vector at the end to avoid repeating red hues at both ends of the palette. From the resulting plot we can see that sample SL_d3072_I achieved the best coverage with 25% of the targeted genome achieving at least 1000X coverage and 75% of the genome acheiving greater than 800X coverage. Sample M_d3068_A appears to have the worst coverage overall with around 50% of the targeted genome covered up to 700X. Sample SB_d3072_A while achieving good coverage over all, has poor coverage for a greater proportion of the genome with 6% of the targeted space covered only up to 200-250X.
# change the colours in our Plots
colorRamp <- rainbow(1200)[1:1050]
covBars(seqCovMatrix2, colour=colorRamp)
As with the majority of ggplot2 functions covBars() can accept additional ggplot2 layers to alter the plot as long as they are passed as a list. Try using what you know to alter the plot we created above to more closely resemble that of the manuscript. Specifically this will entail the following:
Make the changes listed above to the GenVisR plot, the final output should look like the plot below.
This Rscript file contains the correct answer.