Introduction to loss of heterozygosity plots
We’ve gone through visualizations of point mutations and copy number changes using GenVisR, another type of genomic alteration that is often usefull to visualize are “Loss of Heterozygosity” events. In a diploid cell there are pairs of chromosomes each containing a single copy of the genome. These pairs each come from haploid gametes and are slightly different from each other leading to heterozygosity throughout much of the genome. Situations can arise however where this inherit heterozygosity of the genome is loss, commonly this is through deletions of a parental copy within a chromosome region also known as hemizygosity. Viewing deletions however will not give a complete picture of LOH as events can arise that will lead to copy-neutral LOH, for example if a parental copy was deleted but the then remaining copy underwent an amplification. In this section we will use the GenVisR function lohSpec() created specifically for viewing loh events.
How lohSpec works
The function lohSpec() works by using a sliding window approach to calculate the difference in the variant allele fractions (VAF) between heterozygous variants in matched tumor normal samples. Essentially, a window of a specified size (defined by the parameter window_size) will obtain the absolute difference between the tumor VAF and the normal VAF, which is assumed to be .5, for each genomic position within the window’s position. All of these points are then averaged to obtain a measure of LOH within the window. This window will then move forward by a length specified by the parameter step, and again cauclate this absolute mean tumor-normal VAF difference metric. This LOH value across all overlapping windows is averaged.
Introduction to demonstration dataset
For this section we will be starting from a file of variants generated from the variant calling algorithm varscan. These variants originate from the breast cancer cell line HCC1395 aligned to “hg19” and were lightly processed to limit to only events called as “Germline” or “LOH” by varscan. Once we read this file in from the url we’ll need to do some minor reformatting as described below to make it compatiable with lohSpec().
# read in the varscan data
lohData <- read.delim("http://genomedata.org/gen-viz-workshop/GenVisR/HCC1395.varscan.tsv", header=FALSE)
# grab only those columns which are required and name them
lohData <- lohData[,c("V1", "V2", "V7", "V11")]
colnames(lohData) <- c("chromosome", "position", "n_vaf", "t_vaf")
# add a sample column
lohData$sample <- "HCC1395"
# convert the normal and tumor vaf columns to fractions
lohData$n_vaf <- as.numeric(gsub("%", "", lohData$n_vaf))/100
lohData$t_vaf <- as.numeric(gsub("%", "", lohData$t_vaf))/100
# limit to just the genome of interest
lohData <- lohData[grepl("^\\d|X|Y", lohData$chromosome),]
The function lohSpec() requires a data frame of heterozygous variant calls with column names “chromosome”, “position”, “n_vaf”, “t_vaf”, and “sample” as it’s primary input. Above we read in our germline variant calls from varscan and subset the data to just the required columns renaming them with colnames() and addin in a “sample” column. If we look at the the documentation we can see that lohSpec() expects proportions for the VAF columns and not percentages. We coerce these columns into this format by using gsub() to remove the “%” symbol and dividing the resultant numeric value from the call as.numeric() by 100. We also subset our data using grepl() to remove anything that is not a canonical chromosome as the chromosomes in the primary data must match those specified in the genome parameter we will use later.
Creating an initial plot
Like it’s counterpart cnSpec(), lohSpec() needs genomic boundaries to be specified in addition to the primary data in order to ensure that the entire genome is plotted when creating the graphic. This can be done via the parameter genome which expects a character string specifying one of “hg19”, “hg38”, “mm9”, “mm10”, or “rn5”. Alternatively a custom genome can be supplied with the y parameter. These parameters are both identical to their cnSpec() counterparts, please refer to the cnSpec tutorial for a more detailed explanation. Now that we have all of the required data let’s go ahead and make a standard lohSpec() plot.
# Create an inital plot
lohSpec(lohData, genome="hg19")
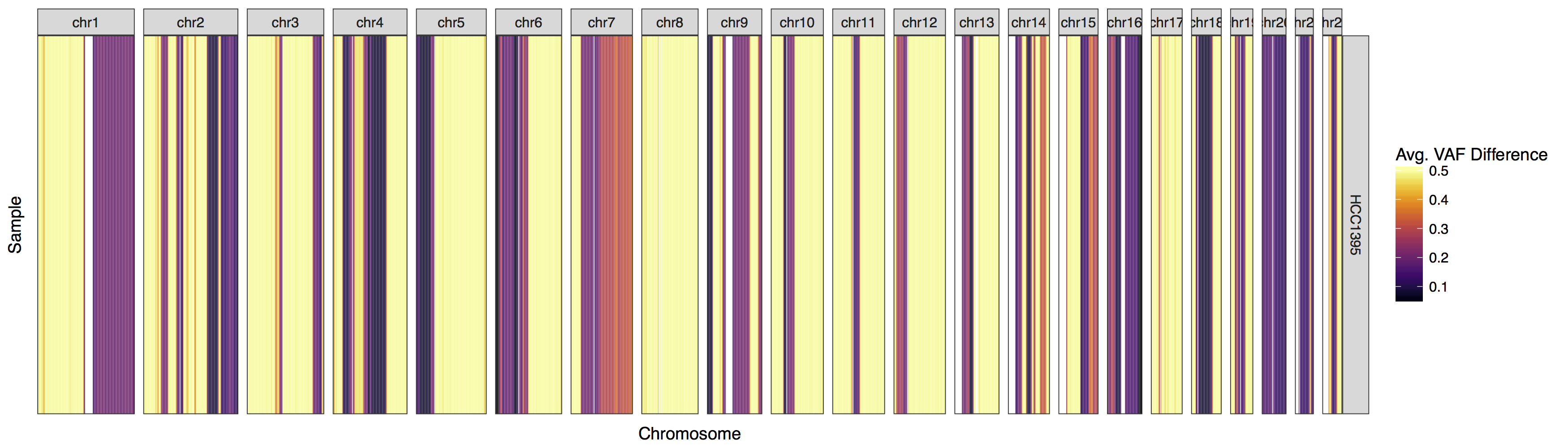
We have our first plot however you might have noticed a warning message along the lines of “Detected values with a variant allele fraction either above .6 or below .4 in the normal. Please ensure variants supplied are heterozygous in the normal!”. Admittedly this warning message is accurate, let’s take a minute to consider why biologically lohSpec() is producing this warning message.
Get a hint!
Refer to the "How lohSpec works" section above
Answer
A heterozygous call in a diploid genome should have a VAF of .5, any deviation from this is either noise due to insufficient coverage, or more problematically indicative that the site is not completely heterozygous. A reasonable explanation could be a focal amplification at that site on one allele. In any case it is inadvisable to use such sites when constructing this type of plot
Now let’s fix the issue and reproduce our plot, note that as expected the warning message has disappeared.
# Obtain variants with a VAF greater than 0.4 and below 0.6.
lohData <- lohData[lohData$n_vaf > 0.4 & lohData$n_vaf < 0.6,]
# run lohSpec
lohSpec(x=lohData)
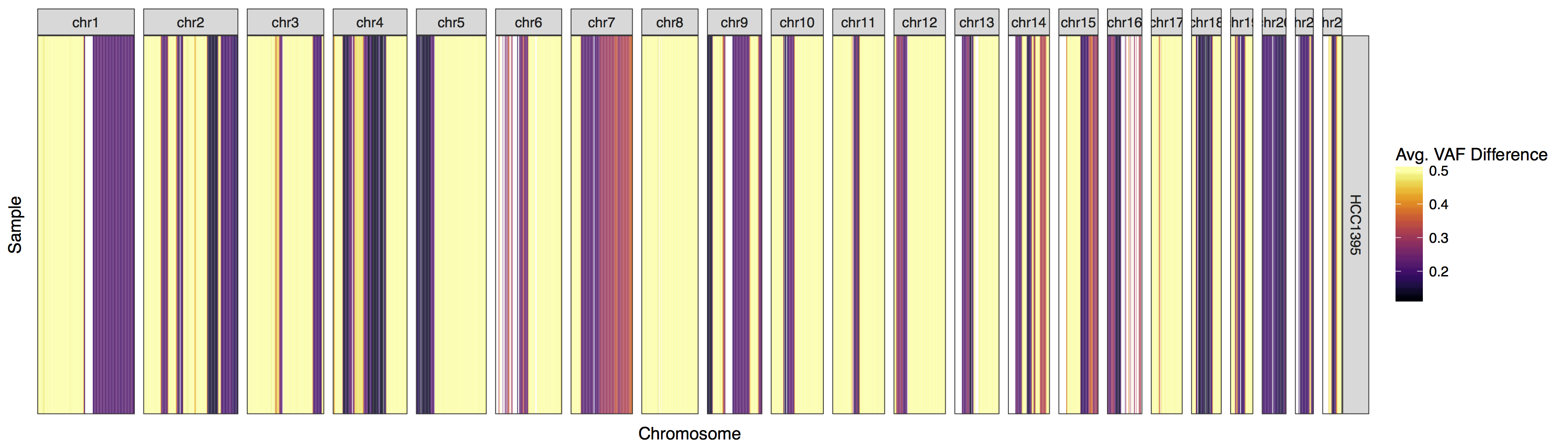
Now that we have a plot let’s go over what it is showing. First off what is a high LOH value. Remember the formula used to calculate LOH for a single variant is |normal vaf - tumor vaf| and that these values are averaged for a given window. Consider this example of LOH then |.5 - 1 | = .5 and |.5 - 0| = .5. Conversely consider this example of non LOH, | .5 - .5 | = 0. So we can see that the closer we get to the value .5 the more evidence there is that an LOH event exists. We have to take noise into account but we can see that the majority of this genome has some sort of LOH, probably not surprising as this data is derived from an imortalized cell line.
Altering the step and window size
At this point it is appropriate to talk about the trade off between speed and accuracy from setting the parameters step and window_size. We have already briefly discussed these parameters and what they do. What has so far gone unsaid is that these parameters really control the amount of smoothing the data undergoes and as such altering one will alter the trade off between the algorithms speed and an accurate representation of the data, this is especially true for the step parameter. We can view the effect of this using the microbenchmark package, increasing the step by a factor of 2/3 will decrease the computation time by almost half. Reasonable defaults have been chosen for the human genome however one should keep these parameters in mind when using a custom genome or when attempting to plot many samples.
# install and load a benchmarking package
# install.packages("microbenchmark")
library(microbenchmark)
# run benchmark tests
microbenchmark(lohSpec(x=lohData, window_size = 2500000, step = 1000000), lohSpec(x=lohData, window_size = 2500000, step = 1500000), times = 5L)
Exercises
There may be situations in which you would want to view only specific regions within the genome instead of the whole genome itself. This can be achieved by supplying a custom genome to the parameter y and making sure your input data is limited to only that region. The gene PTEN is commonly lost in breast cancer, take a look to see if it’s lost in this cell line. Limit your data to only chromosome 10 and use plotLayer to highlight the q23.31 cytogenetic band on which PTEN resides (chr10:89500000-92900000). Your plot should look something like the one below.
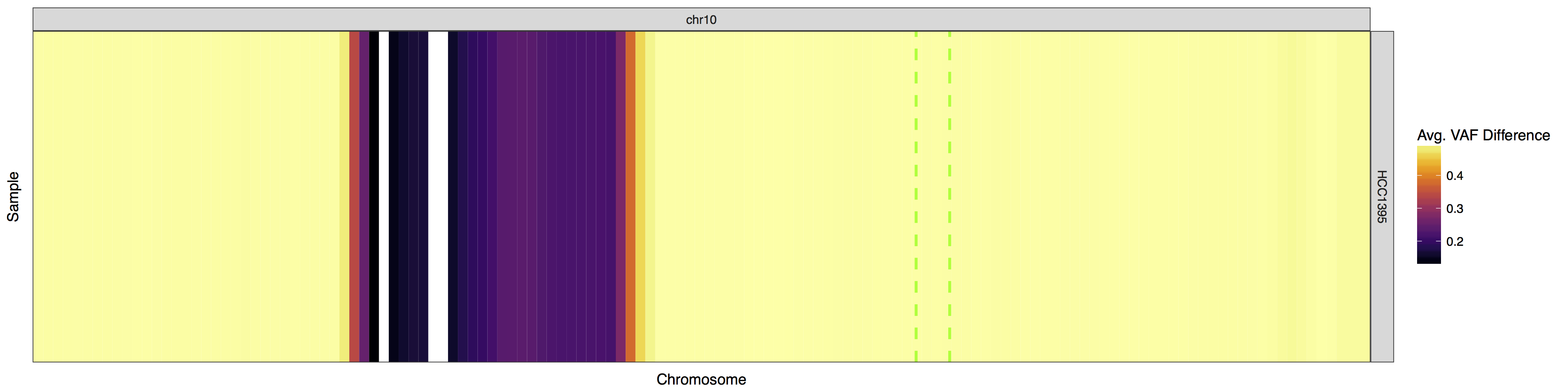
Get a hint!
You'll need to create a custom genome that is only chromosome 10, and you'll need to subset your input data as well.
Get a hint!
Look at the messages lohSpec() outputs, it automatically prepends chr to the input to x so your custom genome will need chr prepended as well
Get a hint!
Try using geom_vline() to highlight q23.1
Answer
The solution is supplied in this file.
At times it may be desireable to alter how the plot looks, lohSpec() has a few helpfull parameters to aid in this but it may also be necessary to add additional plot layers as well via the parameter plotLayer. try to recreate the plot below using a combination of parameters in the lohSpec() documentation and adding additional layers via plotLayer. You will need to alter the plot colours, add a title, and change the facets to take up an equal share of the plot.
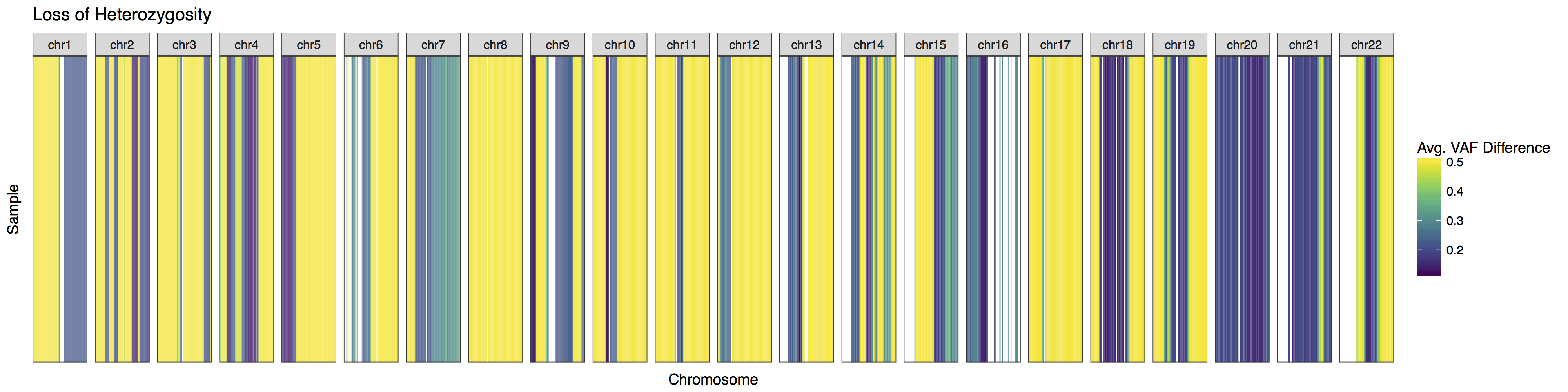
Get a hint!
look at the parameter "colourScheme"
Get a hint!
you will need to add layers for facet_grid() and ggtitle()
Get a hint!
Remember from earlier that when multiple layers are supplied they must be as a list!
Answer
lohSpec(lohData, colourScheme = "viridis", plotLayer=list(facet_grid(.~chromosome, scales="free"), ggtitle("Loss of Heterozygosity")))
When plotting these types of plots, particular care must be taken when dealing with allosomes. The parameter gender is usefull in this regard however try and think why it is necessary to consider for these plots and why lohSpec() does not plot allosomes by default.
Answer
Depending on the gender, the allosomes of a sample could be diploid or haploid, if the later is true LOH could be deceptively displayed when there is not any.