Introduction to liftover tools
A common analysis task is to convert genomic coordinates between different assemblies. Probably the most common situation is that you have some coordinates for a particular version of a reference genome and you want to determine the corresponding coordinates on a different version of the reference genome for that species. For example, you have a bed file with exon coordinates for human build GRC37 (hg19) and wish to update to GRCh38. Many resources exist for performing this and other related tasks. In this section we will go over a few tools to perform this type of analysis, in many cases these tools can be used interchangeably. This post is inspired by this BioStars post (also created by the authors of this workshop).
Download this first
http://hgdownload.soe.ucsc.edu/goldenPath/hg38/liftOver/hg38ToCanFam3.over.chain.gz
Reference assemblies
First let’s go over what a reference assembly actually is. A reference assembly is a complete (as much as possible) representation of the nucleotide sequence of a representative genome for a specific species. These assemblies provide a powerful shortcut when mapping reads as they can be mapped to the assembly, rather than each other, to piece the genome of a new individual together. This has a number of benefits, the most obvious of which is that it is far more effecient than attempting to build a genome from scratch. By its very nature however using this approach means there is no perfect reference assembly for an individual due to polymorphisms (i.e. snps, hla-type, etc.). Furthermore, due to the presence of repetitive structural elements such as duplications, inverted repeats, tandem repeats, etc. a given assembly is almost always incomplete, and is constantly being improved upon. This leads to the publication of new assembly versions every so often such as grch37 (Feb. 2009) and grch38 (Dec. 2013) for the Human Genome Project. It is also important to be aware that different organizations can publish different reference assemblies, for example grch37 (NCBI) and hg19 (UCSC) are identical save for a few minor differences such as in the mitochondria sequence and naming of chromosomes (1 vs chr1). For a nice summary of genome versions and their release names refer to the Assembly Releases and Versions FAQ.
Resources available
There are many resources available to convert coordinates from one assemlby to another. We will go over a few of these. However, below you will find a more complete list. The UCSC liftOver tool is probably the most popular liftover tool, however choosing one of these will mostly come down to personal preference.
-
UCSC liftOver: This tool is available through a simple web interface or it can be downloaded as a standalone executable. To use the executable you will also need to download the appropriate chain file. Each chain file describes conversions between a pair of genome assemblies. Liftover can be used through Galaxy as well. There is a python implementation of liftover called pyliftover that does conversion of point coordinates only.
-
NCBI Remap: This tool is conceptually similar to liftOver in that it manages conversions between a pair of genome assemblies but it uses different methods to achieve these mappings. It is also available through a simple web interface or you can use the API for NCBI Remap.
-
The Ensembl API: The final example I described above (converting between coordinate systems within a single genome assembly) can be accomplished with the Ensembl core API. Many examples are provided within the installation, overview, tutorial and documentation sections of the Ensembl API project. In particular, refer to these sections of the tutorial: ‘Coordinates’, ‘Coordinate systems’, ‘Transform’, and ‘Transfer’.
-
Assembly Converter: Ensembl also offers their own simple web interface for coordinate conversions called the Assembly Converter.
-
rtracklayer: For R users, Bioconductor has an implementation of UCSC liftOver in the rtracklayer package.
-
CrossMap: A standalone open source program for convenient conversion of genome coordinates (or annotation files) between different assemblies. It supports most commonly used file formats including SAM/BAM, Wiggle/BigWig, BED, GFF/GTF, VCF. CrossMap is designed to liftover genome coordinates between assemblies. It’s not a program for aligning sequences to reference genome. Not recommended for converting genome coordinates between species.
-
Flo: A liftover pipeline for different reference genome builds of the same species. It describes the process as follows: “align the new assembly with the old one, process the alignment data to define how a coordinate or coordinate range on the old assembly should be transformed to the new assembly, transform the coordinates.”
Using UCSC liftOver
Sex linkage was first discovered by Thomas Hunt Morgan in 1910 when he observed that the eye color of Drosophila melanogaster did not follow typical Mendelian inheritance. This was discovered to be caused by the white gene located on chromosome X at coordinates 2684762-2687041 for assembly dm3. Let’s use UCSC liftOver to determine where this gene is located on the latest reference assembly for this species, dm6. First navigate to the liftOver site at https://genome.ucsc.edu/cgi-bin/hgLiftOver and set both the original and new genomes to the appropriate species, “D. melanogaster”.
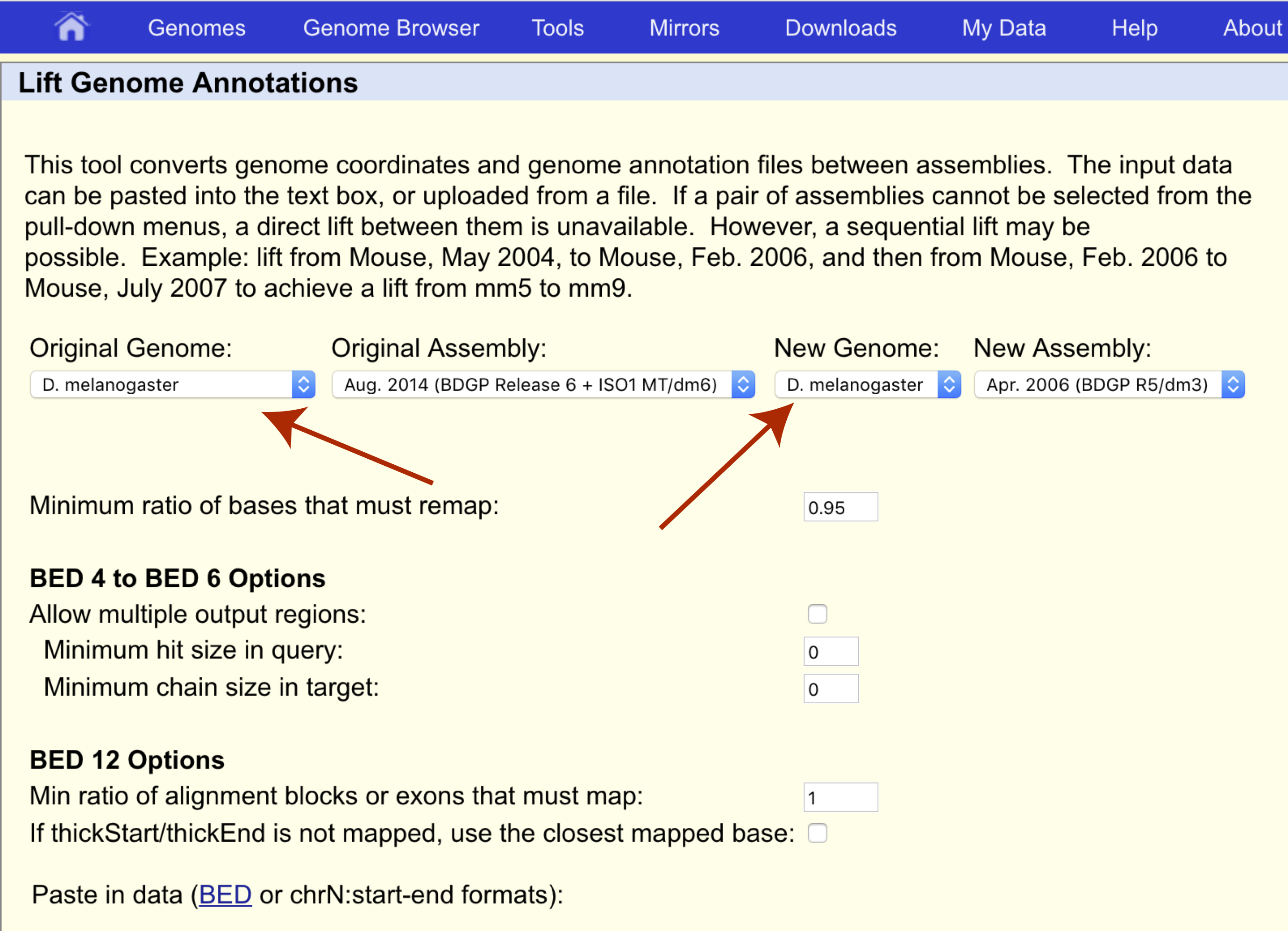
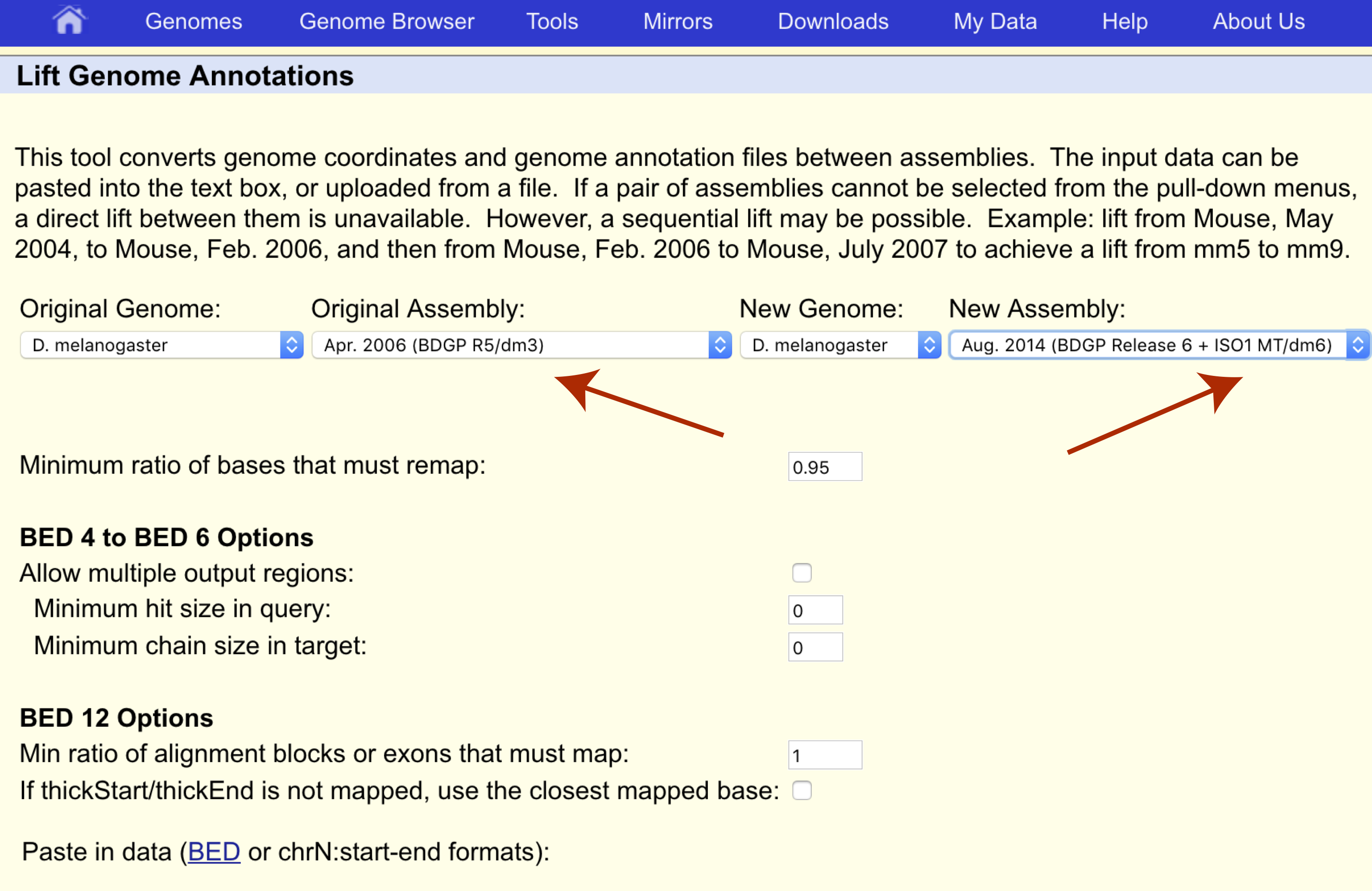
We want to transfer our coordinates from the dm3 assembly to the dm6 assembly so let’s make sure the original and new assemblies are set appropriately as well.
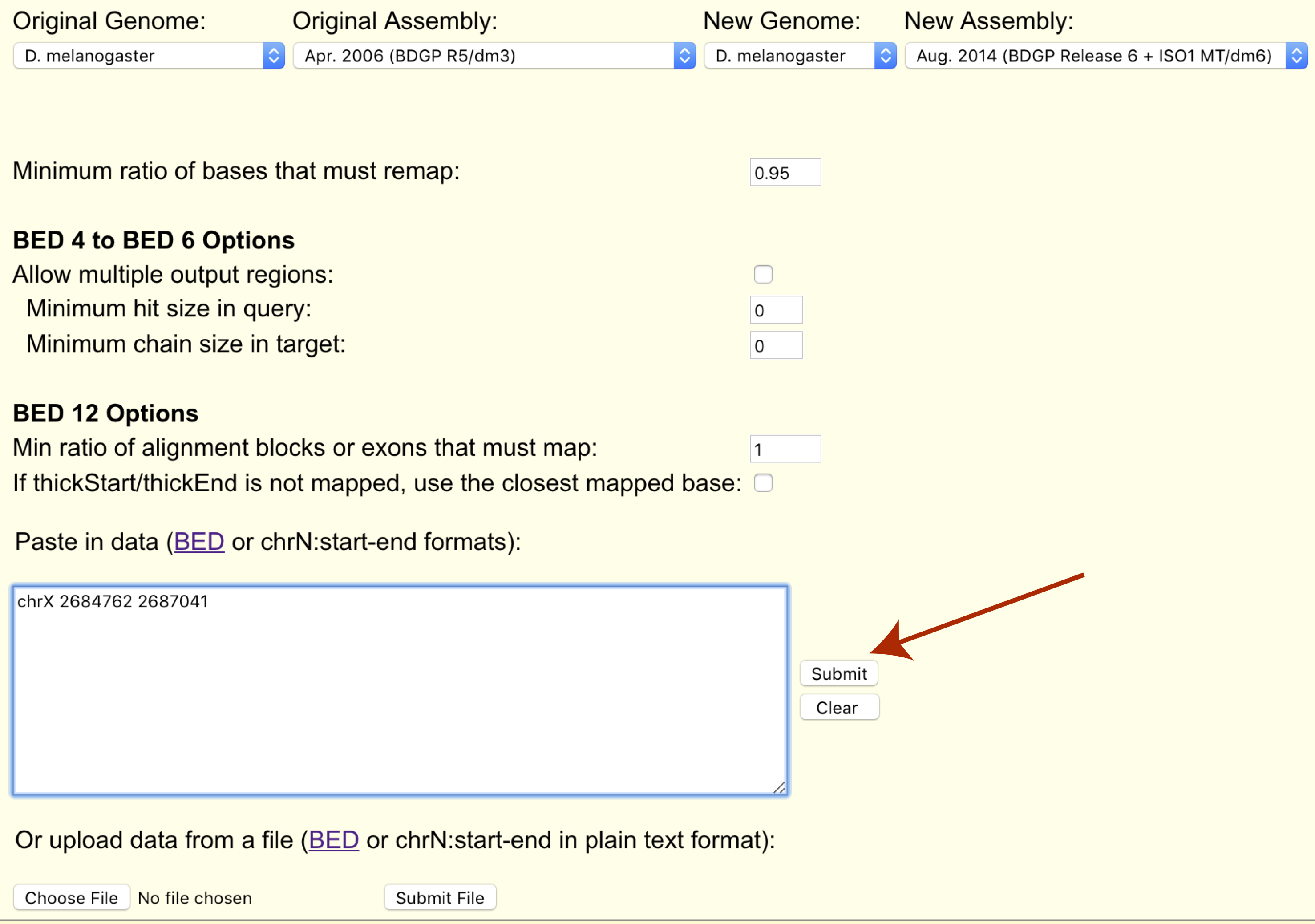
Finally we can paste our coordinates to transfer or upload them in bed format (chrX 2684762 2687041).
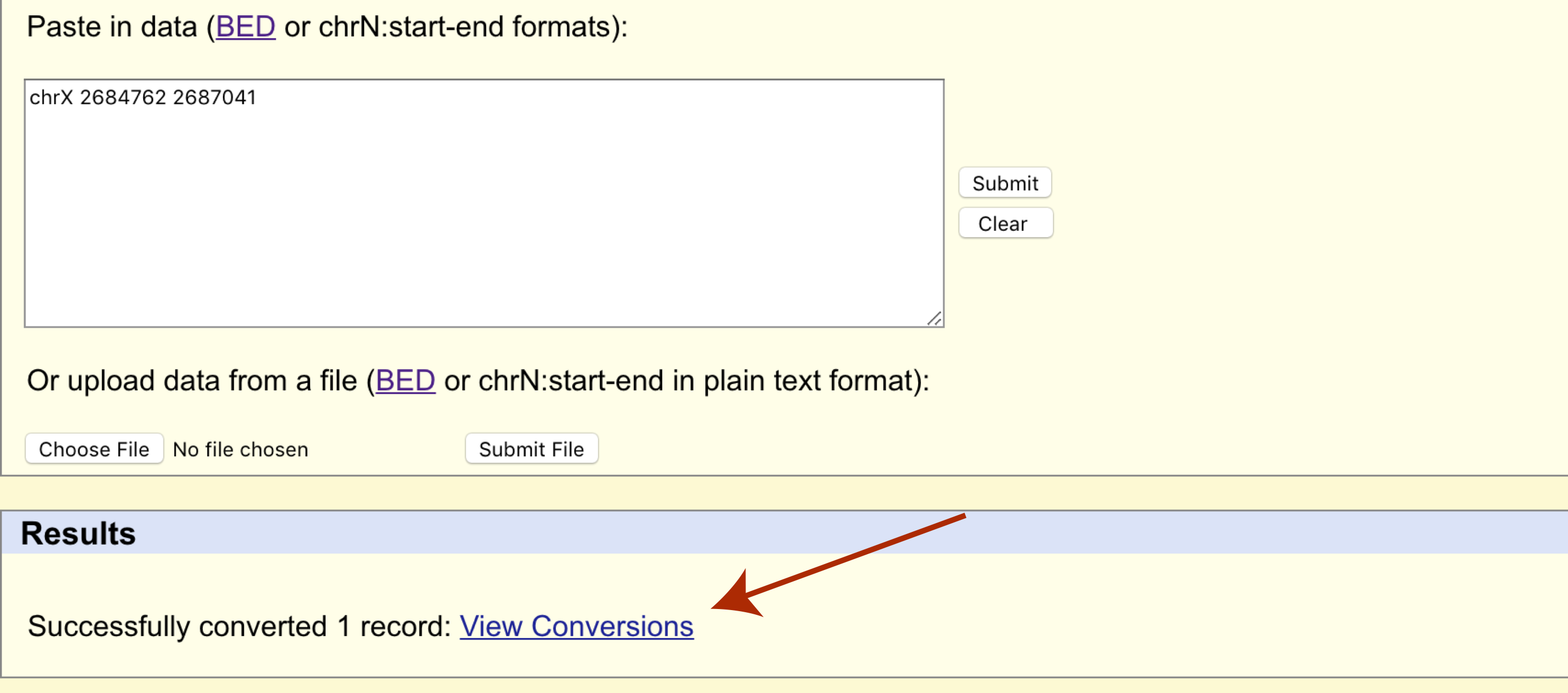
The page will refresh and a results section will appear where we can download the transferred cordinates in bed format.
Try and compare the old and new coordinates in the UCSC genome browser for their respective assemblies, do they match the same gene?
Answer
Yes, both coordinates match the coding sequence for the w gene from transcript CG2759-RA
Using rtracklayer
In another situation you may have coordinates of a gene and wish to determine the corresponding coordinates in another species. This is a common situation in evolutionary biology where you will need to find coordinates for a conserved gene across species to perform a phylogenetic analysis. Let’s use the rtracklayer package on bioconductor to find the coordinates of the H3F3A gene located at chr1:226061851-226071523 on the hg38 human assembly in the canFam3 assembly of the canine genome. To start install the rtracklayer package from bioconductor, as mentioned this is an R implementation of the UCSC liftover.
# install and load rtracklayer
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("rtracklayer", version = "3.8")
library(rtracklayer)
The function we will be using from this package is liftover() and takes two arguments as input. The first of these is a GRanges object specifying coordinates to perform the query on. This class is from the GenomicRanges package maintained by bioconductor and was loaded automatically when we loaded the rtracklayer library. The second item we need is a chain file, which is a format which describes pairwise alignments between sequences allowing for gaps. The UCSC website maintains a selection of these on it’s genome data page. Navigate to this page and select “liftOver files” under the hg38 human genome, then download and extract the “hg38ToCanFam3.over.chain.gz” chain file.
Next all we need to do is to create our GRanges object to contain the coordinates chr1:226061851-226071523 and import our chain file with the function [import.chain()]. We can then supply these two parameters to liftover().
# specify coordinates to liftover
grObject <- GRanges(seqnames=c("chr1"), ranges=IRanges(start=226061851, end=226071523))
# import the chain file
chainObject <- import.chain("hg38ToCanFam3.over.chain")
# run liftOver
results <- as.data.frame(liftOver(grObject, chainObject))
How many different regions in the canine genome match the human region we specified?
Answer
210, these return the ranges mapped for the corresponding input element
Exercises
Try to perform the same task we just complete with the web version of liftOver, how are the results different?
Answer
Both methods provide the same overall range, however using rtracklayer is not simplified and contains multiple ranges corresponding to the chain file.