Posts
My thoughts and ideas
Welcome to the blog
My thoughts and ideas
COSMIC, the Catalogue Of Somatic Mutations In Cancer, hosted by the Wellcome Trust Sanger Institute, is one of the largest and most comprehensive resources for exploring the impact of somatic mutations in human cancer. It is the product of a massive expert data curation effort, presenting data from thousands of publications and many important large scale cancer genomics datasets. COSMIC acts as the main portal for at least four important projects or resources: COSMIC - the Catalogue Of Somatic Mutations In Cancer, the Cancer Gene Census, the Cell Lines Project and COSMIC-3D. Academic users can use and download (with registration) the data for free, so long as they do not re-distribute the data, and agree to the license terms. For-profit users must pay a fee to download COSMIC. These resources provide access to a very rich set of data, visualizations, and interpretations for understanding cancer mutations and cancer genes.
To browse COSMIC you can simply navigate to the main page and search for a gene, cancer type, mutation, etc in the search box. To illustrate we will explore the results for a single gene. Type BRAF in the search interface and hit enter. At the time of writing, this search term returned results for 1 gene, 826 mutations, 49 cancers, 226 samples, 1575 Pubmed citations, and 0 studies (see results tabs). Note that COSMIC contains results for almost 300,000 tested samples and over 50,000 mutations in BRAF.
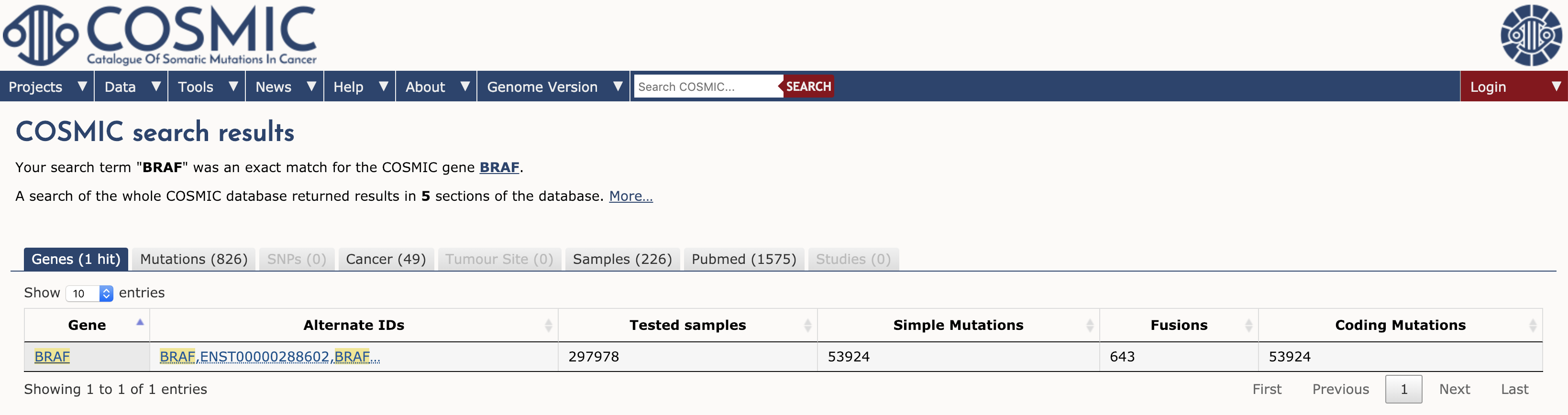
If we select the BRAF result, COSMIC returns a detailed page that provides: gene summaries, links to other COSMIC resources (e.g., Census genes, Hallmark genes, etc), external links, drug resistance, tissue distribution, genome browser view, mutation distribution, variants, and references. We will look at few of these sections. First, let’s look at the Overview section. Along the top of this section there are several useful icons. The ‘Census gene’ icon tells us that BRAF is a known cancer gene according to the Gene Census (see below). The next three icons tell us that it is also an ‘Expert curated gene’, that mouse insertional mutagenesis experiments support that BRAF is a cancer gene, and finally that BRAF is a ‘Cancer Hallmark’ gene. After these icons are many more details about BRAF including coordinates, synonyms, link to COSMIC-3D (see below), and more.
Next, let’s examine the Gene view. The histogram of mutation (substitution) frequency shows a very dramatic “hotspot” of mutations at position 600 (e.g., p.V600E). Mouse over this part of the histogram to see details. This is a very well-known driver mutation in multiple types of cancer.
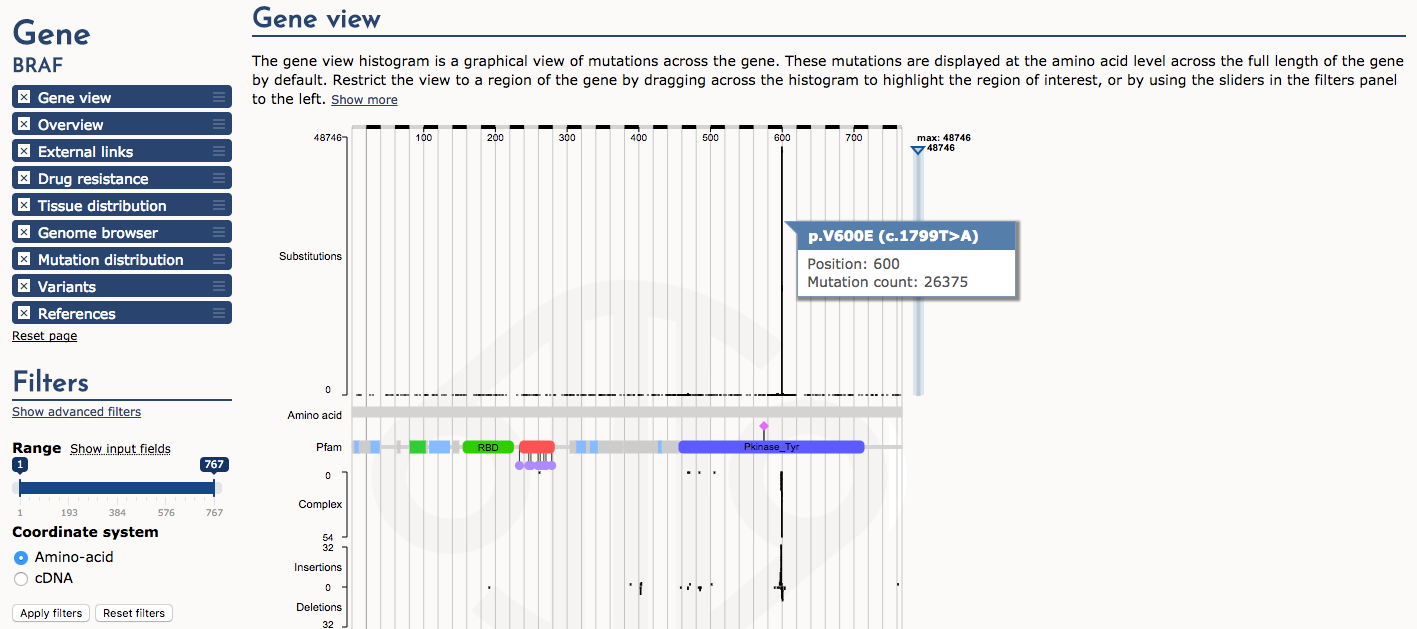
What part (domain) of the BRAF protein is affected by the p.V600E mutation?
The protein tyrosine kinase domain (Pfam)
Finally, navigate to the ‘Tissue distribution’ section. Sort the table by ‘Point mutations’ -> ‘% Mutated’. Notice that cancers of the thyroid and skin (e.g., melanoma) are by far the most consistently mutated at the BRAF gene locus (note NS means not specified). A subset of samples also display copy number variation (CNV) gains and up-regulated expression. In general certain predominanly mutated genes tend to be associated with cancers of certain origins. However, there are many exceptions to this statement and some genes (e.g., TP53) are widely mutated in many different cancer types.
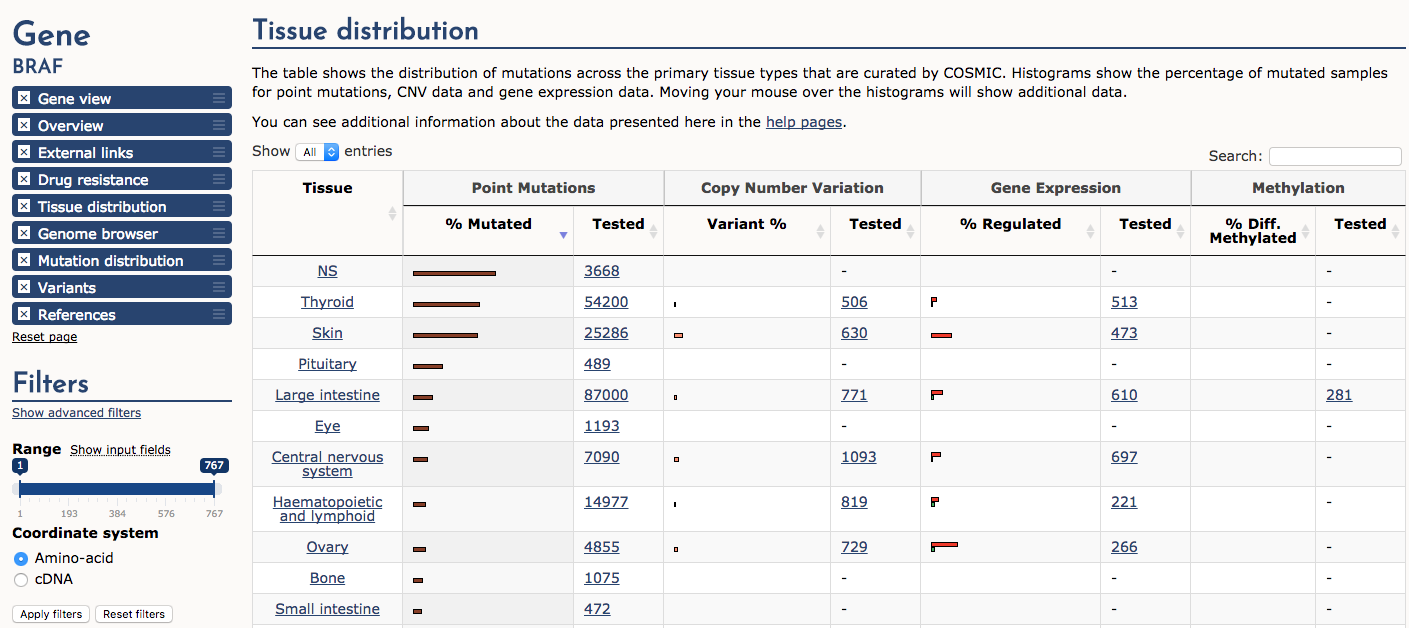
What cancer tissue type is most commonly affected by BRAF over-expression?
Approximately 15% of ovarian cancers are affected by BRAF over-expression
The Cancer Gene Census (CGC) is an ongoing effort to catalogue those genes for which mutations (somatic or germline) have been causally implicated in cancer. The original census and analysis was published in Nature Reviews Cancer by Futreal et al. 2004 but it continues to receive regular updates. The CGC is widely regarded as a definitive list of cancer genes (tumor suppressors and oncogenes). Navigate to the Cancer Gene Census from the dropdown ‘Projects’ menu available on any COSMIC page. This page is broken into three main sections: Cancer Gene Census, Breakdown, and Abbreviations. In the first section, a simple table of all Cancer Gene Census genes is displayed.
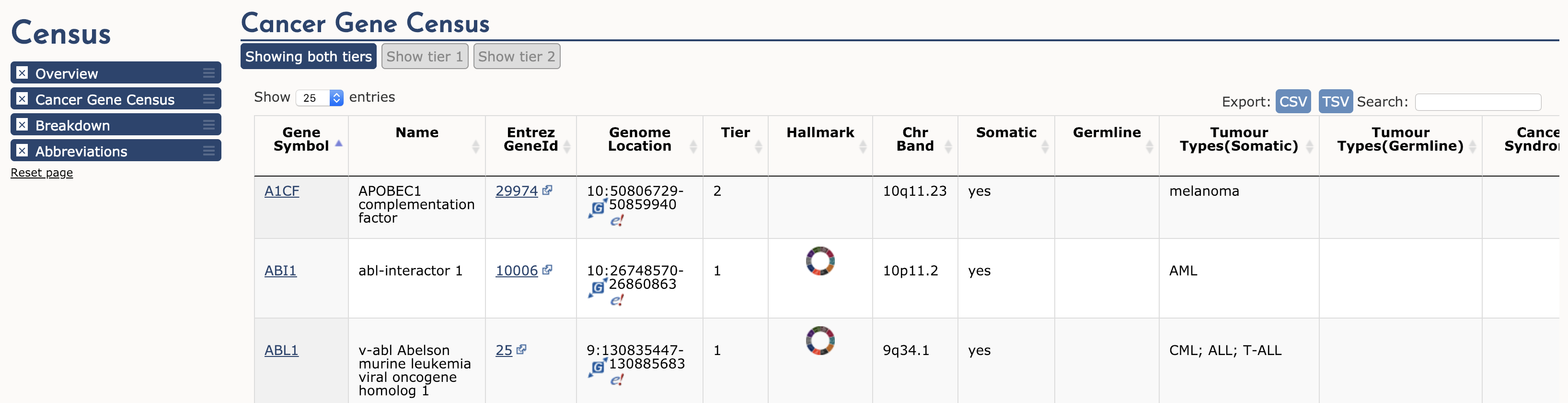
To illustrate, lets examine ABL1 (the third gene in the table). The first four columns provide the gene’s descriptive name, links to its COSMIC and Entrez gene pages, and genomic region with links to COSMIC and Ensembl Browser views. Other columns tell us that ABL1 is located on chromosome band 9q34.1, known to be somatically mutated in CML, ALL, and T-ALL. It acts as an oncogene and as a gene fusion partner. In fact, ABL1 is one part of perhaps the most famous cancer fusion, BCR-ABL, the product of the Philadelphia chromosome rearrangement. This fusion defines and drives nearly all cases of CML, and its discovery lead to one of the most successful examples of targeted therapy (Imatinib). To learn more about ABL1’s role in cancer select the ‘Census Hallmark’ icon. A ‘Hallmark’ is a reference to the seminal paper by Hanahan and Weinberg (2000), updated in 2011, in which they proposed that all cancers share six (now ten) common traits (hallmarks) that explain the transformation of normal cells into malignant cancer cells. Cancer Gene Census curators have attemped to assign each cancer gene in the census to one or more of these Hallmarks. The graphic shows which hallmarks are promoted (green bars) or suppressed (blue bars) by ABL1. In this case, ABL1 is thought to promote proliferative signalling, change of cellular energetics, genome instability, angiogenesis, invasion and metastasis, and suppress programmed cell death.
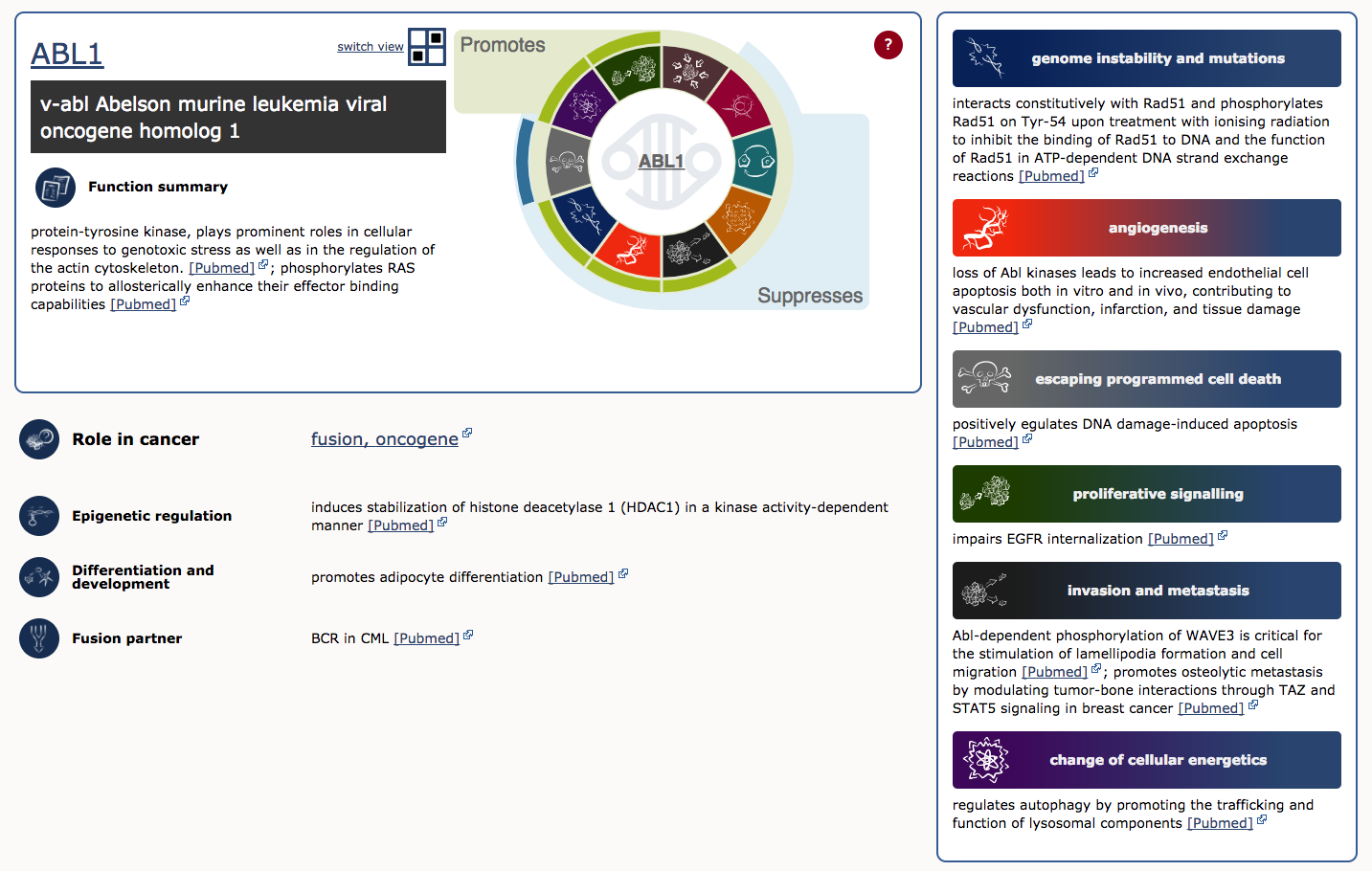
How many tumor supressor genes (TSGs) and oncogenes are there in CGC?
At time of writing there were 315 TSGs and 315 oncogenes in the CGC. Interestingly, there are 72 genes that function as both oncogene and TSG.
Note: You will need to register with COSMIC to download the complete Cancer Gene Census. You could then load this file in R, and use linux commandline, or some other approach to determine the list of TSGs and oncogenes currently documented in the CGC.